
Strategic Wins



- Sanofi and QuantHealth team up to advance AI-powered digital twins and clinical trial
- simulations to accelerate drug development
Tel Aviv & Cambridge, MA – October 1st, 2025 — QuantHealth, a pioneer in AI-driven clinical trial simulation, announced a strategic investment from Sanofi Ventures, the venture capital arm of global healthcare leader Sanofi. The investment will accelerate QuantHealth’s efforts to bring scalable, patient-level simulations and digital twin technologies to the forefront of drug development, bringing its total funds raised to $30 million.
QuantHealth’s platform enables pharmaceutical companies to virtually simulate clinical trials using real-world data and advanced AI models. By generating millions of patient-level digital twins, the platform predicts trial outcomes and optimizes protocol design, aiming to improve trial success rates, timelines, and cost efficiency.
The investment has further catalyzed an enterprise strategic relationship that supports Sanofi’s focus on integrating advanced AI and digital technologies to drive transformation across its R&D ecosystem. As part of the investment, Cris De Luca, Partner at Sanofi Ventures, will join as an observer on QuantHealth’s Board of Directors.
“We’re proud to welcome Sanofi Ventures as an investor and strategic ally in our journey,” said Orr Inbar, CEO and Co-founder of QuantHealth. “Sanofi is at the forefront of digital transformation in pharma, and this relationship will help us scale our impact and bring more predictive, AI-driven approaches to clinical development.”
“QuantHealth has the potential to transform how clinical trials are designed and optimized,” said Cris De Luca, Partner at Sanofi Ventures. “Their approach to leveraging digital twins and real-world data is advancing the next generation of R&D, and I look forward to supporting the team.”
“At Sanofi, we are building an AI-first organization, and engaging with innovators like QuantHealth is central to our strategy,” said Emmanuel Frenehard, Chief Digital Officer at Sanofi. “Their platform represents an important opportunity to reimagine clinical trial design through simulation, and we’re excited to explore this capability together.”
This investment and enterprise relationship underscores a shared commitment to advancing drug development through the responsible use of artificial intelligence in life sciences.
About QuantHealth
QuantHealth is an AI-driven clinical trial simulation company that helps pharmaceutical companies
design faster, more successful trials using real-world data and predictive modeling. With access to over 350 million patient records and proprietary AI algorithms, QuantHealth’s platform simulates trials at scale to optimize protocols, reduce risk, and accelerate timelines.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi, investing globally in early-stage biotech and digital health companies aligned with Sanofi’s mission to chase the miracles of science. Areas of focus include immunology, oncology, rare diseases, vaccines, and digital innovation.



- Sanofi receives exclusive rights to commercialize losmapimod in all territories outside the U.S.; Fulcrum retains full U.S. commercialization rights
- Fulcrum will receive an upfront payment of $80.0 million, and is eligible to receive $975.0 million in potential milestones, plus royalties on ex-U.S. product sales; parties will share future global development costs 50:50
- Conference call and webcast scheduled for 8:00 a.m. ET today to discuss the collaboration and other recent corporate developments, in conjunction with the first quarter 2024 financial results
CAMBRIDGE, Mass., May 13, 2024 (GLOBE NEWSWIRE) -- Fulcrum Therapeutics, Inc.® (Fulcrum) (Nasdaq: FULC), a clinical-stage biopharmaceutical company focused on developing small molecules to improve the lives of patients with genetically defined rare diseases, today announced that it has entered into a collaboration and license agreement with Sanofi (Nasdaq: SNY) for the development and commercialization of losmapimod, an oral small molecule being investigated for the treatment of facioscapulohumeral muscular dystrophy (FSHD). Under the collaboration and license agreement, Sanofi obtains exclusive commercialization rights for losmapimod outside of the U.S.
The collaboration and license agreement combines Fulcrum’s expertise in FSHD with Sanofi’s global reach and unparalleled commitment to treating patients with rare diseases. Losmapimod is currently being evaluated in a global Phase 3 clinical trial for the treatment of FSHD, a chronic and progressive genetic muscular disorder that is characterized by significant muscle cell death and fat infiltration into muscle tissue. Results from ReDUX4, the Phase 2 clinical trial evaluating losmapimod for the treatment of FSHD, demonstrated a slowing of disease progression and improved muscle health. Fulcrum expects to report topline data from REACH, the global Phase 3 clinical trial, in the fourth quarter of 2024. Following positive data from the Phase 3 trial, Fulcrum and Sanofi plan to submit marketing applications in the U.S., Europe, Japan, and other geographies.
“Sanofi is a proven leader in developing therapeutics for rare neuromuscular diseases and is the ideal partner to maximize the opportunity and reach of losmapimod outside the U.S.,” said Alex C. Sapir, Fulcrum’s president and chief executive officer. “This deal aligns with our core strategy, allowing Fulcrum to remain focused on preparations for commercialization of losmapimod in the U.S., while leveraging Sanofi’s exceptional global commercial capabilities and established infrastructure in key markets around the world. We are excited about the potential to provide the first approved treatment for FSHD patients, and we look forward to working with Sanofi to bring losmapimod to patients globally.”
“This partnership provides an exciting opportunity to expand Sanofi’s rare disease franchise and deliver the first approved FSHD treatment to patients with the strength and reach of our commercial organization,” said Burcu Eryilmaz, Sanofi’s Global Head of Rare Diseases. “Losmapimod has shown meaningful clinical benefits that underscore the disease-modifying potential and opportunity to address the high unmet need for a safe and effective drug that slows disease progression. With a deep commitment to bringing hope and new treatment options to patients, we look forward to working closely with Fulcrum as losmapimod advances through late-stage development.”
Per the terms of the agreement, Fulcrum will receive an upfront payment of $80.0 million and is eligible to receive up to an additional $975.0 million in specified regulatory and sales-based milestones, along with tiered escalating royalties starting in the low-teens on annual net sales of losmapimod outside the U.S. In addition, Fulcrum and Sanofi will equally share future global development costs.
Conference Call and Webcast
Individuals may register for the conference call by clicking the link here. Once registered, participants will receive dial-in details and unique PIN which will allow them to access the call. An audio webcast will be accessible through the Investor Relations section of Fulcrum’s website at www.fulcrumtx.com or by clicking here. Following the live webcast, an archived replay will also be available.
About Losmapimod
Losmapimod is a selective p38α/β mitogen activated protein kinase (MAPK) inhibitor. Fulcrum exclusively in-licensed losmapimod from GSK following Fulcrum’s discovery of the role of p38α/β inhibitors in the reduction of DUX4 expression and an extensive review of known compounds. Results reported from the Phase 2 ReDUX4 trial demonstrated a slowing of disease progression and improved function, including positive impacts on upper extremity strength and functional measures supporting losmapimod’s potential to be a transformative therapy for the treatment of FSHD. Although losmapimod had never previously been explored in muscular dystrophies, it had been evaluated in more than 3,600 subjects in clinical trials across multiple other indications, with no safety signals attributed to losmapimod. Losmapimod has been granted U.S. Food and Drug Administration (FDA) Fast Track designation and Orphan Drug Designation for the treatment of FSHD. Losmapimod is currently being evaluated in a Phase 3 multi-center randomized, double-blind, placebo-controlled, 48-week parallel-group study in people with FSHD (NCT05397470).
About FSHD
FSHD is a serious, rare, progressive and debilitating disease for which there are no approved treatments. It is characterized by fat infiltration of skeletal muscle leading to muscular atrophy involving primarily the face, scapula and shoulders, upper arms, and abdomen. Impact on patients includes profound decreases in the ability to perform activities of daily living, loss of upper limb function, loss of mobility and independence and chronic pain. FSHD is one of the most common forms of muscular dystrophy and has an estimated patient population of 30,000 in the United States alone.
About Fulcrum Therapeutics
Fulcrum Therapeutics is a clinical-stage biopharmaceutical company focused on developing small molecules to improve the lives of patients with genetically defined rare diseases in areas of high unmet medical need. Fulcrum’s two lead programs in clinical development are losmapimod, a small molecule in development for the treatment of facioscapulohumeral muscular dystrophy (FSHD), and pociredir (formerly known as FTX-6058), a small molecule designed to increase expression of fetal hemoglobin and in development for the treatment of sickle cell disease (SCD). Fulcrum uses proprietary technology to identify drug targets that can modulate gene expression to treat the known root cause of gene mis-expression. For more information, visit www.fulcrumtx.com and follow us on Twitter/X (@FulcrumTx) and LinkedIn.
Forward-Looking Statements
This press release contains “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995 that involve substantial risks and uncertainties. All statements, other than statements of historical facts, contained in this press release are forward-looking statements, including express or implied statements regarding Fulcrum’s collaboration and license agreement with Sanofi and receipt of the upfront payment thereunder; its ability to receive the milestone and royalty payments thereunder and achieve benefits therefrom; timing of data from REACH and its ability to support submission of marketing applications for losmapimod; and Fulcrum’s ability to deliver an FDA-approved therapy for FSHD patients; among others. The words “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “target,” “will,” “would” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. Any forward-looking statements are based on management’s current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in, or implied by, such forward-looking statements. These risks and uncertainties include, but are not limited to, risks associated with Fulcrum’s ability to continue to advance its product candidates in clinical trials; initiating and enrolling clinical trials on the timeline expected or at all; obtaining and maintaining necessary approvals from the FDA and other regulatory authorities; replicating in clinical trials positive results found in preclinical studies and/or earlier-stage clinical trials of losmapimod, pociredir and any other product candidates; obtaining, maintaining or protecting intellectual property rights related to its product candidates; managing expenses; managing executive and employee turnover, including integrating a new CMO; and raising the substantial additional capital needed to achieve its business objectives, among others. For a discussion of other risks and uncertainties, and other important factors, any of which could cause Fulcrum’s actual results to differ from those contained in the forward-looking statements, see the “Risk Factors” section, as well as discussions of potential risks, uncertainties, and other important factors, in Fulcrum’s most recent filings with the Securities and Exchange Commission. In addition, the forward-looking statements included in this press release represent Fulcrum’s views as of the date hereof and should not be relied upon as representing Fulcrum’s views as of any date subsequent to the date hereof. Fulcrum anticipates that subsequent events and developments will cause Fulcrum’s views to change. However, while Fulcrum may elect to update these forward-looking statements at some point in the future, Fulcrum specifically disclaims any obligation to do so.
Contact:
Chris Calabrese
LifeSci Advisors, LLC
ccalabrese@lifesciadvisors.com
917-680-5608


Collaboration will focus on co-development of unique datasets to understand health in everyday life
January 10, 2022 06:00 AM Eastern Standard Time
SAN MATEO, Calif.--(BUSINESS WIRE)--Evidation, the company creating new ways to measure and improve health in everyday life, is expanding its decade-long collaboration with Sanofi to build upon their joint real-world data initiatives. This new phase will focus on the co-development of unique datasets to develop and validate new measures of health and wellness.
Evidation’s collaboration with Sanofi has delivered groundbreaking results to date, with over 20 studies conducted across 10 therapeutic areas, including diabetes and Type 2 Inflammation, more than 500,000 patients reached, and four studies published. This continued collaboration will further the work Evidation and Sanofi have pioneered to translate person-generated health data into quantified clinical and economic outcomes, a key priority for both companies.
“After nearly a decade of working with Sanofi, we are proud to expand this collaboration agreement to advance the role that real-world data and analysis can play in better understanding health and disease,” said Christine Lemke, Evidation co-founder and co-CEO. “Sanofi has led the way in garnering insights from real-world data in R&D and we’re excited to advance our work together into its next decade."
Sanofi and Evidation announced a prior expansion of their work together in 2017, in addition to Sanofi’s investment in Evidation in the same year.
“Real-world evidence is critical to help us better understand the patient’s health and wellness journey outside of traditional healthcare visits,” said Arnaud Robert, Executive Vice President, Chief Digital Officer, Sanofi. “Through our expanded collaboration with Evidation, we can further our ambition to transform the practice of medicine by connecting more closely with patients and citizens, expanding our geographic capabilities, and increasing diversity to better represent the global population.”
This announcement comes as biopharmaceutical companies, regulators, and payers are working to develop new guidelines on how real-world data should be incorporated into the development and approval of therapeutics. Evidation and Sanofi will continue to contribute to this conversation through similar industry-leading research.
The Evidation network is made up of more than 4.4 million individuals across all 50 states, representing 9 out of every 10 U.S. ZIP codes, allowing organizations like Sanofi access to a highly engaged, diverse population and privacy-conscious research platform.
ABOUT EVIDATION
Evidation measures health in everyday life and enables anyone to participate in ground-breaking research and health programs. Built upon a foundation of user privacy and control over permissioned health data, Evidation’s platform is trusted by millions of individuals—generating data with unprecedented speed, scale, and rigor. We partner with leading healthcare companies to understand health and disease outside the clinic walls. Guided by our mission to enable and empower everyone to participate in better health outcomes, Evidation is working to bring people individualized, proactive, and accessible healthcare—faster. Founded in 2012, Evidation is headquartered in California with additional offices around the globe. To learn more, visit evidation.com, or follow us on Twitter @evidation.
Contacts
MEDIA CONTACT
Matt Miller
press@evidation.com


Partnership to create new treatment delivery options for people facing serious diseases
BOSTON, MA—February 10, 2021—i2O Therapeutics, developers of a platform for oral delivery of traditionally injectable biological drugs, announced today a research collaboration with Sanofi to investigate the oral delivery of Sanofi's Nanobody®-based medicines, which are currently administered through intravenous or subcutaneous injections.
Nanobodies - proprietary therapeutic proteins based on camelid derived immunoglobulin single variable domains - have potential uses in the treatment of a range of serious and life-threatening diseases and are being developed in many therapeutic areas including inflammation, hematology, immuno-oncology, oncology and rare diseases. The research collaboration between i2O Therapeutics and Sanofi will explore a new oral route of administering nanobodies.
“Our mission at i2O Therapeutics is to develop safe and effective oral formulations of therapies traditionally limited to injections and we are excited to partner with Sanofi to advance this mission,” said Ravi Srinivasan, co-founder and director of i2O Therapeutics.
“i2O’s ionic liquid platform opens new opportunities to orally deliver biologics, and nanobodies represent an exciting application of this platform,” said Samir Mitragotri, co-founder of i2O Therapeutics.
i2O Therapeutics announced seed funding in April 2020, which was led by Sanofi Ventures, the corporate venture capital arm of Sanofi, and JDRF T1D Fund. The company also announced a strategic investment from Colorcon Ventures, the corporate venture capital fund of Colorcon, Inc. in December 2020.
About i2O Therapeutics
i2O Therapeutics is a biotechnology company developing safe and effective oral formulations of therapies traditionally limited to injections. Using an innovative ionic liquid technology, this platform leverages the benefits of protecting the drug cargo while also transiently enhancing permeation across the epithelial lining when administered orally. i2O is focused on creating the next generation of oral peptide and protein-based therapies. Visit us at www.i2OBio.com.
About Sanofi
Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions. With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe.
Contact
Lauren Arnold
MacDougall
larnold@macbiocom.com
781-235-3060


- Kymera to receive $150 million upfront with more than $2 billion in potential milestones plus royalty payments
- Kymera to retain option during clinical development to participate equally in US cost and profit sharing
CAMBRIDGE, Mass. (July 9, 2020) – Kymera Therapeutics Inc. today announced the company has entered into a multi-program strategic collaboration with Sanofi (NASDQ: SNY) to develop and commercialize first-in-class protein degrader therapies targeting IRAK4 in patients with immune-inflammatory diseases. The companies will also partner on a second earlier stage program. Kymera will receive $150 million in cash upfront and may receive more than $2 billion in potential development, regulatory and sales milestones, as well as significant royalty payments. Kymera retains the option to participate in US development and commercialization for both programs. This includes the ability to participate equally in the costs, profits and losses after opt-in, and to co-promote partnered products in the US.
“This is an important collaboration for both companies and for the field of targeted protein degradation,” said Nello Mainolfi, Ph.D., co-founder, President and CEO of Kymera Therapeutics. “Kymera is becoming a fully integrated biotechnology company advancing a pipeline of novel therapies with the potential to transform treatment paradigms. We are excited to partner with Sanofi, an organization with world-class drug development and commercialization capabilities, to ensure maximal patient impact from two of our programs across multiple disease indications, while enabling Kymera to invest in key strategic areas to realize the broad potential of protein degrader therapies.”
Under terms of the collaboration, Sanofi will make an upfront payment of $150 million in cash to Kymera for global rights to develop its small molecule IRAK4 protein degraders in inflammation and immunology indications, and a second earlier stage undisclosed program. IRAK4 is believed to play a key role in multiple immune-inflammatory diseases, including hidradenitis suppurativa, atopic dermatitis and rheumatoid arthritis. Kymera will advance the IRAK4 program through Phase 1 clinical trials; Sanofi will assume clinical development and commercialization responsibilities thereafter. Sanofi will lead all clinical development activities for the second program. Kymera will have the option to participate in the development of both programs in the US during clinical development. Kymera will retain global rights to its IRAK4 program in oncology indications.
IRAK4 is a key protein involved in inflammation mediated by the activation of toll-like receptors (TLRs) and IL-1 receptors (IL-1Rs). While TLR and IL-1R signaling via IRAK4 is involved in the normal immune response, aberrant activation of these pathways is the underlying cause of multiple immune-inflammatory conditions. In pre-clinical studies, Kymera has shown oral daily administration of an IRAK4 degrader can lead to complete knockdown of IRAK4 in skin and immune cells in higher species and is well tolerated. Data presented at the most recent annual meetings of the American College of Rheumatology and the European Hidradenitis Suppurativa Foundation showed potent anti- inflammatory activity in both in vitro and in vivo preclinical models."
“Targeted protein degrada on is an exci ng modality. Kymera has developed an incredible drug discovery engine producing protein degraders with compelling and dieren ated pharmacology against targets that, to date, have not been op mally addressed with other therapeu c modali es,” said John Reed, Global Head of Research & Development at Sano. “We are excited to partner with the Kymera team to advance a new genera on of rst-in-class therapies with the poten al to eliminate underlying drivers of disease.”
Aquilo Partners, L.P. acted as Financial advisor to Kymera on this transac on.
# # #
About Kymera Therapeutics
Kymera Therapeutics is a biotechnology company pioneering a transformative new approach to treating previously untreatable diseases. The company is advancing the field of targeted protein degradation, accessing the body’s innate protein recycling machinery to degrade dysregulated, disease-causing proteins. Kymera’s Pegasus targeted protein degradation platform harnesses the body’s natural protein recycling machinery to degrade disease-causing proteins, with a focus on un-drugged nodes in validated pathways currently inaccessible with conventional therapeutics.
Kymera is accelerating drug discovery with an unmatched ability to target and degrade the most intractable of proteins, and advance new treatment options for patients. For more information, visit www.kymeratx.com.
About Sanofi
Sanofi is dedicated to supporting people through their health challenges. We are a global pharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions. With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe. Sanofi, empowering life. For more information, visit www.sanofi.com.


PARIS and NEW YORK – November 20, 2019 - Sanofi announced today an enterprise- wide collaboration with health care technology company Aetion that will integrate Sanofi’s real-world data platform, DARWIN, with the Aetion Evidence Platform® with the objective of advancing more efficient use of real-world evidence (RWE), facilitating regulatory-grade studies with deep transparency, and unlocking access to new real-world data.
Both companies have invested in RWE platforms, recognizing the pressing need for accurate, fast, and cost-effective research and the important role RWE could play in meeting this need. Sanofi’s DARWIN compiles and analyzes de-identified data from hundreds of millions of patients across disease states, while Aetion’s platform analyzes real-world data to produce transparent, rapid, and scientifically validated answers about the effectiveness, safety, and value of drugs. By combining these platforms, Sanofi is seeking to elevate its capabilities in conducting regulatory-grade analytics, opening new doors for the development and application of medical treatments.
“Today marks another important step in Sanofi’s digital transformation,” said Bernard Hamelin, MD, MSc, MBA, Global Head of Medical Evidence Generation, Sanofi. “By integrating these platforms, we strive to make faster, more informed decisions with the potential to lead to first-in-class and best-in-class treatments that could change the practice of medicine.”
Real-world evidence offers a view of clinical practice outside of the experimental setting, providing an opportunity to inform clinical trial development and supplement trial data with evidence of actual product use in the health care system.
“Our work with Sanofi further validates the value and potential for real-world evidence in drug development,” said Carolyn Magill, Chief Executive Officer of Aetion. “Our companies share a common goal of using the best available data to get the right treatment to the right patient as quickly and efficiently as possible.”
This collaboration between Sanofi and Aetion demonstrates leadership during a critical time. Real-world evidence is expected to play a key role in transforming the health care ecosystem, with the U.S. Food and Drug Administration (FDA) recently prioritizing efforts to incorporate RWE as a companion to clinical trial data to aid in regulatory decision making. The FDA will release its draft RWE guidance before the end of 2020.
About Aetion
Aetion is a health care technology company that delivers real-world evidence for life sciences companies, payers, at-risk providers, and regulatory agencies. The Aetion Evidence Platform analyzes data from the real world to produce transparent, rapid, and scientifically validated answers on treatments, costs, and outcomes. Founded by Harvard Medical School faculty with decades of experience in epidemiology and health outcomes research, Aetion informs health care's most critical decisions — what works best, for whom, and when — to guide treatment development, commercialization, and payment innovation into health care's modern era. Aetion is based in New York City, and backed by investors including New Enterprise Associates (NEA), Flare Capital Partners, Lakestar, Town Hall Ventures, McKesson Ventures, Sanofi Ventures, Amgen Ventures, UCB, and Horizon Health Services, Inc. Learn more at aetion.com, and follow us at @aetioninc.
About Sanofi
Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions.
With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe.
Sanofi, Empowering Life
Sanofi Media Relations Contact
Anna Robinson
Tel.: +33 (0)1 53 77 46 46
mr@sanofi.com
Aetion Media Relations Contact
202.792.7200
press@aetion.com
Sanofi Investor Relations Contact
George Grofik
Tel.: +33 (0)1 53 77 45 45
ir@sanofi.com
Sanofi Forward-Looking Statements
This press release contains forward-looking statements as defined in the Private Securities Litigation Reform Act of 1995, as amended. Forward-looking statements are statements that are not historical facts. These statements include projections and estimates and their underlying assumptions, statements regarding plans, objectives, intentions and expectations with respect to future financial results, events, operations, services, product development and potential, and statements regarding future performance. Forward-looking statements are generally identified by the words “expects”, “anticipates”, “believes”, “intends”, “estimates”, “plans” and similar expressions. Although Sanofi’s management believes that the expectations reflected in such forward- looking statements are reasonable, investors are cautioned that forward-looking information and statements are subject to various risks and uncertainties, many of which are difficult to predict and generally beyond the control of Sanofi, that could cause actual results and developments to differ materially from those expressed in, or implied or projected by, the forward-looking information and statements. These risks and uncertainties include among other things, the uncertainties inherent in research and development, future clinical data and analysis, including post marketing, decisions by regulatory authorities, such as the FDA or the EMA, regarding whether and when to approve any drug, device or biological application that may be filed for any such product candidates as well as their decisions regarding labelling and other matters that could affect the availability or commercial potential of such product candidates, the absence of guarantee that the product candidates if approved will be commercially successful, the future approval and commercial success of therapeutic alternatives, Sanofi’s ability to benefit from external growth opportunities, to complete related transactions and/or obtain regulatory clearances, risks associated with intellectual property and any related pending or future litigation and the ultimate outcome of such litigation, trends in exchange rates and prevailing interest rates, volatile economic conditions, the impact of cost containment initiatives and subsequent changes thereto, the average number of shares outstanding as well as those discussed or identified in the public filings with the SEC and the AMF made by Sanofi, including those listed under “Risk Factors” and “Cautionary Statement Regarding Forward-Looking Statements” in Sanofi’s annual report on Form 20-F for the year ended December 31, 2018. Other than as required by applicable law, Sanofi does not undertake any obligation to update or revise any
forward-looking information or statements.



Without clinical trials, new medicine may never make it from the research lab to patients in need. These carefully designed studies can provide important data that include proper dosage, benefit to patients, and potential side effects.
There is a growing challenge, however, in finding appropriate participants, especially for treatments that target highly specific conditions affecting narrower patient populations. Right now, there are more than 40,000 clinical studies recruiting patients in the U.S. alone, with some requiring thousands of participants, each of whom must meet precise criteria to join. So it’s not surprising that 80% of these important studies are delayed due to recruitment problems, according to a study by the Center for Information and Study on Clinical Research Participation (CISCRP).
Unfortunately, those delays mean it can take longer for innovative new medicines to be studied and approved, leaving patients to wait years for new treatment options. To tackle this growing problem, Sanofi is taking a digital approach to clinical trials, partnering with Science 37, a clinical research services and technology company based in California.
Leveraging mobile technology and telemedicine capabilities, this new approach will allow Sanofi to develop “site-less” or decentralized clinical trials that are more patient friendly: easier for them to access, and eliminating many of the common impediments to participation. Using digital technologies to streamline finding and retaining participants for the entire length of a study has the potential to reduce the time required for a typical trial by at least 30%, according to Science 37.


“After years invested in the lab on an innovative treatment, the clinical trials are where we finally obtain and analyze the relevant data that will let us understand how well a new treatment will benefit patients,” said Lionel Bascles, Global Head of Clinical Sciences and Operations of Sanofi. “With digital clinical trials we can get and analyze the data on how a new medicine works in the real world a lot sooner, which means patients get the medicines they need sooner.”
Going digital also eliminates a number of other hurdles to patient participation, including the most significant: geography.
Most people are eager to participate in relevant trials – 87% of patients want to do so, the CISCRP study found. Yet, 70% of potential participants live more than two hours away from the nearest study center. Because most clinical trials require patients to travel to those centers for regular tests and observations, sometimes several times each week for the duration of the trial, this distance is another challenge to patient access.
Science 37’s approach allows patients to be monitored and report to researchers via an Apple iPhone equipped with the company’s NORA® technology. Qualified study participants are provided with the phone, a data plan and any other sensors or connected devices needed for the trial, along with the medicines being researched. Participants can reach study staff at any time via the mobile device, while also remaining under the care of their local health care professionals. Mobile nurses are also sent to the participant’s home to provide services like blood draws when needed, and nearby hospitals or clinics are engaged for scans or other tests that require specialized equipment.
The patient’s data are sent securely to researchers who can immediately access information that would otherwise have to be collected by medical personnel through face-to-face interactions at study centers. This platform can also remind patients to take their study medications at the proper time, and let researchers know if participants are adhering to the study requirements.
“Our decentralized clinical trial model addresses critical shortcomings of traditional clinical trials, such as enrolling and retaining appropriate patients. Whether you live near a major research institution, or in a remote area, we make participation possible,” said Noah Craft, CEO of Science 37. “By utilizing a patient’s home in lieu of a physical trial site, we remove the burden of travel for those too sick or remote and provide access to qualified individuals who want to volunteer for a study but cannot because of geographic limitations.”
The Science 37 platform will also help engage patients who would normally not participate in clinical trials, “so our data will much more closely track the diversity of the population,” Bascles said. “In addition to reducing the burden for patients, decentralized clinical trials are far more likely to keep patients engaged for the full length of the trial, increasing the relevance and the acceptability of the data by regulators.”
Sanofi’s agreement with Science 37 covers use of its Metasite™ model and NORA technology across the U.S. with plans to expand internationally in the future. By eliminating months of searching for patients and long travel time to study sites, virtual clinical trials could reduce total trial time by as much as two years.
Partnering with Science 37 is the most recent strengthening of the relationship with Sanofi, which began last October when Sanofi’s venture capital fund, Sanofi-Genzyme BioVentures, made a minority investment in Science 37.
“Science 37 has a great track record, and they are smart and forward-thinking about developing the science around clinical trials that leverage digital technologies,” said Heather Bell, Global Head of Digital and Analytics for Sanofi. “As part of the scope of our digital strategy, we have expanded the scope of the venture fund to include digital investments, and Science 37 was our first investment since that change and we’re very excited about it.”


SEATTLE and SOUTH SAN FRANCISCO – October 16, 2014 Immune Design Corp. (NASDAQ: IMDZ), a clinical-stage immunotherapy company, today announced that it has entered into a broad collaboration for the development of a herpes simplex virus (HSV) immune therapy with Sanofi Pasteur, the vaccines division of Sanofi (EURONEXT: SAN and NYSE: SNY).
Sanofi Pasteur and Immune Design will each contribute product candidates to the collaboration: Sanofi Pasteur will contribute HSV-529, a clinical-stage replication-defective HSV vaccine product candidate, and Immune Design will contribute G103, its preclinical trivalent vaccine product candidate. The collaboration will explore the potential of various combinations of agents, including leveraging Immune Design’s GLAASTM platform, with the goal to select the best potential immune therapy for patients.
The two companies will develop the products jointly through Phase 2 clinical trials, at which point Sanofi Pasteur intends to continue development of the most promising candidate and be responsible for commercialization. Sanofi Pasteur will bear the costs of all preclinical and clinical development, with Immune Design providing a specific formulation of GLA from the GLAAS platform at its cost through Phase 2 studies. Immune Design will be eligible to receive future milestone and royalty payments on any product developed from the collaboration; other financial terms of the agreement have not been disclosed.
“Instead of being limited to a single approach, the companies are joining forces and combining multiple cutting-edge technologies with the goal to develop the most effective and safe immunotherapy to address HSV infection, a significant unmet medical need,” said Carlos Paya, M.D., Ph.D., President and Chief Executive Officer of Immune Design. “With other clinical and preclinical GLAAS-based product candidates in development, both with partners and internally at Immune Design, I believe this new collaboration continues to demonstrate the productivity and broad applicability of this platform.”
About G103 and GLAAS
G103 is a trivalent vaccine candidate consisting of recombinantly-expressed viral proteins adjuvanted with a specific formulation from Immune Design’s GLAAS platform. The combination of a novel molecular toll-like receptor 4 (TLR4) agonist with rationally selected antigens is designed to boost pre-existing T cells and trigger a broad antibody response, allowing for prophylactic and therapeutic immunization.
The GLAAS platform works in vivo and is based on a small synthetic molecule called GLA, which stands for glucopyranosyl lipid adjuvant. GLA selectively binds to the TLR4 receptor and causes potent activation of dendritic cells (DCs) leading to the production of cytokines and chemokines that drive a Th1-type immune response. When GLA is accompanied by an antigen and injected into a patient, the combination is taken up by DCs and leads to the production and expansion of immune cells called CD4 T helper lymphocytes with a Th1 phenotype. These CD4 T cells play a key role in boosting pre-existing cytotoxic T cells that are specific to the same antigen; and providing help to other immune cells, including B lymphocytes that are the precursor to antibodies, and natural killer cells that are also important in the overall immune response. Immune Design believes that GLAAS- based product candidates have the potential to target multiple types of cancer, as well as infectious, allergic and autoimmune diseases. Product candidates leveraging GLAAS’ core technology have now
been evaluated in over 1000 subjects in Phase 1 and Phase 2 trials.
About Immune Design
Immune Design (NASDAQ: IMDZ) is a clinical-stage immunotherapy company employing next- generation in vivo approaches to enable the body’s immune system to fight disease. The company’s technologies are engineered to activate the immune system’s natural ability to create and/or expand antigen-specific cytotoxic T cells, while enhancing other immune effectors, to fight cancer and other chronic diseases. Immune Design’s three on-going immuno-oncology clinical programs are the product of its two synergistic discovery platforms: ZVexTMand GLAASTM, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Immune Design Cautionary Note Regarding Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend", “believe” and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design’s expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include statements regarding efforts to develop products under the collaboration, the potential receipt of milestone and royalty payments and the potential to develop new therapeutics. Factors that may cause actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design’s filings with the U.S. Securities and Exchange Commission (the “SEC”), including the "Risk Factors" section of Immune Design’s Quarterly Report on Form 10-Q filed with the SEC on September 8, 2014 and in any subsequent filings with the SEC. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
# # #
For Immune Design:
Media Contact Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact Robert H. Uhl
Westwicke Partners
robert.uhl@westwicke.com
858-356-5932
CAMBRIDGE, Mass. and SEATTLE and SOUTH SAN FRANCISCO, Calif., Aug. 7, 2014 (GLOBE NEWSWIRE) -- Sanofi
(EURONEXT:SAN) (NYSE:SNY) and Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company, today announced that they have entered into a licensing agreement for use of Immune Design's GLAASTM discovery platform to develop therapeutic agents to treat a selected food allergy.
The incidence of food allergies is increasing worldwide in both developed and undeveloped countries, and especially in children.1 Globally, experts believe 220-250 million people may suffer from food allergies.2,3 In the United States alone, as many as 15 million people have food allergies,4 with allergic reactions resulting in an emergency room visit every three minutes and averaging more than 200,000 emergency room visits per year.5
"This is an exciting time in the area of immunology research, and our relationship with Immune Design is a great example of how Sanofi has changed our approach to R&D," said Kurt Stoeckli, vice president and head of Global Bio Therapeutics Organization, Sanofi. "With this partnership, we are able to tap into breakthrough science that holds great potential to transform how food allergies are treated, and the lives of those people affected. This kind of innovation is central to our new approach."
Under terms of the agreement, Immune Design has granted Sanofi an exclusive license to discover, develop and commercialize products to treat a selected food allergy. The company has received an undisclosed upfront payment and will be eligible to receive development and commercialization milestones totaling US $168 million, as well as tiered royalties on sales of approved products.
"Our fourth agreement for the use of the GLAAS platform further demonstrates the broad applicability of this approach not only in cancer and infectious diseases, but now in allergic diseases as well," said Stephen Brady, chief business officer at Immune Design. "Due to the immune dysfunction leading to allergic diseases, GLAAS' mechanism of action is well suited to correct the imbalance, allowing for the potential of new therapeutics in the targeted indication that currently uses century-old technologies. We are pleased that Sanofi has decided to develop products for this often life-threatening and growing food allergy."
Under an existing collaborative research arrangement, Sanofi and Immune Design have generated a large set of preclinical data demonstrating that certain formulations within GLAAS, when given prophylactically or therapeutically, can shift the immune responses in a way that may result in significant protection and reduction from allergy symptoms.
About Sanofi
Sanofi, an integrated global healthcare leader, discovers, develops and distributes therapeutic solutions focused on patients' needs. Sanofi has core strengths in the field of healthcare with seven growth platforms: diabetes solutions, human vaccines, innovative drugs, and consumer healthcare, emerging markets, animal health and the new Genzyme. Sanofi is listed in Paris (EURONEXT:SAN) and in New York (NYSE:SNY).
About GLAAS
Immune Design's GLAAS platform works in vivo and is based on a small synthetic molecule called GLA, which stands for glucopyranosyl lipid adjuvant. GLA selectively binds to the TLR4 receptor and causes potent activation of dendritic cells (DCs) leading to the production of cytokines and chemokines that drive a Th1-type immune response. When GLA is accompanied by an antigen and injected into a patient, the combination is taken up by DCs and leads to the production and expansion of immune cells called CD4 T helper lymphocytes with a Th1 phenotype. These CD4 T cells play a key role in boosting pre- existing CTLs that are specific to the same antigen; and providing help to other immune cells, including B lymphocytes that are the precursor to antibodies, and natural killer cells that are also important in the overall immune response. Immune Design believes that GLAAS product candidates have the potential to target multiple types of cancer, as well as infectious, allergic and autoimmune diseases. GLAAS-based product candidates have now been evaluated in over 1000 subjects in Phase 1 and Phase 2 trials demonstrating an acceptable safety profile and efficacy.
About Immune Design
Immune Design (Nasdaq:IMDZ) is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to create tumor-specific cytotoxic T cells, while enhancing other immune effectors, to fight cancer and other chronic diseases. Immune Design's three on-going Immuno-oncology clinical programs are the product of its two synergistic discovery platforms: DCVexTMand GLAASTM, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute, respectively. Immune Design has offices in Seattle, Washington and South San Francisco, California. For more information, visit www.immunedesign.com.
Sanofi Forward-Looking Statements
This press release contains forward-looking statements as defined in the Private Securities Litigation Reform Act of 1995, as amended. Forward-looking statements are statements that are not historical facts. These statements include projections and estimates and their underlying assumptions, statements regarding plans, objectives, intentions and expectations with respect to future financial results, events, operations, services, product development and potential, and statements regarding future performance. Forward-looking statements are generally identified by the words "expects", "anticipates", "believes", "intends", "estimates", "plans" and similar expressions. Although Sanofi's management believes that the expectations reflected in such forward-looking statements are reasonable, investors are cautioned that forward-looking information and statements are subject to various risks and uncertainties, many of which are difficult to predict and generally beyond the control of Sanofi, that could cause actual results and developments to differ materially from those expressed in, or implied or projected by, the forward-looking information and statements. These risks and uncertainties include among other things, the uncertainties inherent in research and development, future clinical data and analysis, including post marketing, decisions by regulatory authorities, such as the FDA or the EMA, regarding whether and when to approve any drug, device or biological application that may be filed for any such product candidates as well as their decisions regarding labelling and other matters that could affect the availability or commercial potential of such product candidates, the absence of guarantee that the product candidates if approved will be commercially successful, the future approval and commercial success of therapeutic alternatives, the Group's ability to benefit from external growth opportunities, trends in exchange rates and prevailing interest rates, the impact of cost containment policies and subsequent changes thereto, the average number of shares outstanding as well as those discussed or identified in the public filings with the SEC and the AMF made by Sanofi, including those listed under "Risk Factors" and "Cautionary Statement Regarding Forward-Looking Statements" in Sanofi's annual report on Form 20-F for the year ended December 31, 2013. Other than as required by applicable law, Sanofi does not undertake any obligation to update or revise any forward-looking information or statements.
Immune Design Cautionary Note Regarding Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend", "believe" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include statements regarding the receipt of milestone and royalty payments, the potential to develop new therapeutics and the potential of any future products to prevent and reduce allergy symptoms. Factors that may cause actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
References
- "Food Allergy - A Rising Global Health Problem," World Allergy Week 2013. 8-14 April 2013. http://www.worldallergy.org/UserFiles/file/WorldAllergyWeek2013final.pdf. Accessed online, July 28, 2014.
- Mills EN, Mackie AR, Burny P, Beyer K, Frewer L et al. "The prevalence, cost and basis of food allergy across Europe." Allergy 2007; 62:717-722.
- Fiocchi A, Sampson HA. "Food Allergy", Section 2.5, in WAO White Book on Allergy, Pawankar R, Canonica GW, Holgate ST, and Lockey RF, editors (Milwaukee, Wisconsin: World Allergy Organization, 2011), pp. 47-53.
- National Institute of Allergy and Infectious Diseases, National Institutes of Health. Report of the NIH Expert Panel on Food Allergy Research. 2006. Accessed online, July 25, 2014. http://www.niaid.nih.gov/topics/foodallergy/research/pages/reportfoodallergy.aspx
- 5. Clark S, Espinola J, Rudders SA, Banerji, A, Camargo CA. Frequency of US emergency department visits for food-
related acute allergic reactions. J Allergy ClinImmunol. 2011; 127(3):682-683.
CONTACT:
Amy BA, Ph.D.
Sanofi Global R&D Communications
Amy.Ba@sanofi.com
Tel: 646-207-4935
Julie Rathbun
Rathbun Communications (Immune Design)
julie@rathbuncomm.com
Tel: 206-769-9219
Latest News

Financing led by new investor, Deep Track Capital with participation from Janus Henderson Investors and Marshall Wace among other investors
Proceeds will support accelerated clinical advancement of company’s pipeline, including lead program DT-101, an AMPA receptor potentiator designed to address significant unmet need in major depressive disorder
NEWTON, Mass., & CARDIFF, United Kingdom -- (BUSINESS WIRE)-- Draig Therapeutics, a clinical-stage biopharmaceutical company developing transformative, best-in-class neuropsychiatric therapies, today announced the closing of an oversubscribed $65 million Series B financing. The round was led by Deep Track Capital with participation from Janus Henderson Investors, Marshall Wace, British Business Bank and Jefferson Life Sciences.
“This oversubscribed financing from an exceptional group of new investors marks an exciting milestone for Draig as we continue to advance our transformative, best-in-class pipeline,” said Ivana Magovčević-Liebisch, PhD, JD, President and Chief Executive Officer of Draig Therapeutics. “Major depressive disorder remains one of the largest unmet needs in medicine. Our next-generation AMPA receptor modulator, DT-101, has shown encouraging safety,
tolerability and target engagement data and displays a pulsatile pharmacokinetic profile which gives us real conviction in its potential. This new financing will enable us to accelerate the
development of our pipeline and bring us closer to our ultimate goal: to restore the brain to a healthier state and enable patients to live their best lives.”
The proceeds will be used to support accelerated clinical development of Draig’s pipeline of highly specific AMPA and GABA receptor modulators which are designed to enable safe,
precise modulation of the major neurocircuits underlying neuropsychiatric disorders. The Company’s lead program, DT-101, is an AMPA receptor potentiator (or positive allosteric
modulator - PAM) designed to address the unmet needs in major depressive disorder (MDD). Phase 2 studies evaluating the effectiveness of DT-101 in patients with MDD are ongoing including a global study for DT-101 as a monotherapy and a U.S. study for DT-101 in an adjunct
setting.
“Draig’s laser-focused execution is a testament to the strength of not only its science, but also its team. In less than two years, the Company has built an international organisation of dedicated individuals, initiated two Phase 2 trials in MDD and advanced its broader pipeline towards the clinic. We are excited to partner with Draig as they continue to advance their best in-
class neuromodulators of validated molecular targets with potential to address significant unmet medical needs," said Rebecca Luse, Managing Director at Deep Track Capital.
About Draig Therapeutics
Draig Therapeutics is a clinical-stage biopharmaceutical company developing transformative, best-in-class neuropsychiatric therapies. Our pipeline of highly specific AMPAR & GABAAR
modulators are designed to enable safe, precise modulation of the major neurocircuits underlying neuropsychiatric disorders. Our lead program, DT-101, is a Phase 2 AMPAR PAM with
best-in-disease potential in major depressive disorder. Working in partnership with patients and their care partners, Draig is transforming the future of neuropsychiatry.
Draig was co-founded by CardiT University and SV Health Investors, which led the seed financing with ICG, and is backed by other leading healthcare venture firms including Access
Biotechnology, Canaan Partners, SR One, Sanofi Ventures and Schroders Capital.
Contacts
Megan Prock McGrath
CTD Comms, LLC
978.800.7468

- Oversubscribed financing led by Ally Bridge Group and co-led by Alpha Wave, with participation from new investors Bain Capital Life Sciences, Fidelity Management & Research Company, T. Rowe Price, a leading sovereign wealth fund, Eventide Asset Management and Velosity Capital, and existing investors
- Proceeds will advance ADVC001, AdvanCell’s novel Lead-212 PSMA-targeted radioligand therapy for metastatic prostate cancer toward Phase 3 development, expand vertically integrated radiopharmaceutical platform and manufacturing infrastructure, and accelerate its growing pipeline of targeted alpha therapies
Boston, USA and Brisbane, Australia – July 15, 2026 – AdvanCell, a clinical-stage radiopharmaceutical company developing innovative targeted alpha therapies for cancer, today announced the closing of an oversubscribed and upsized US $315 million Series D financing.
The financing was led by Ally Bridge Group and co-led by Alpha Wave, alongside new investors Bain Capital Life Sciences, Fidelity Management & Research Company, funds and accounts advised by T. Rowe Price Associates, Inc., a leading sovereign wealth fund, Eventide Asset Management, and Velosity Capital. Existing investors Morningside, Eli Lilly and Company, SV Health Investors, Sanofi Ventures, Abingworth, SymBiosis, Tenmile, Brandon Capital, Piper Heartland, Catalio Capital Management, Proto Axiom, Time BioVentures and other shareholders also participated in the round.
Targeted alpha therapy is entering a new era in oncology, but until now, broader adoption has been limited by isotope supply and manufacturing challenges. AdvanCell has built a vertically integrated platform centered on proprietary Lead-212 technology that combines secure isotope supply, automated manufacturing and scalable production to accelerate the development and commercial delivery of next-generation targeted alpha therapies.
The financing will advance ADVC001 toward Phase 3 clinical development in metastatic prostate cancer, expand AdvanCell's proprietary Lead-212 platform, strengthening isotope supply and expanding U.S. manufacturing infrastructure to support Phase 3 development and future commercial demand, and accelerate AdvanCell’s growing pipeline of targeted alpha therapies.
“This financing marks a transformational milestone for AdvanCell and reflects the conviction of an exceptional investor syndicate in the potential of ADVC001 and the innovation behind our vertically integrated Lead-212 platform,” said Philina Lee, Ph.D., Chief Executive Officer, AdvanCell. “Building on our recent leadership appointments and U.S. expansion, this financing puts us in a strong position to enter our next stage of growth and execution, advancing our lead therapy ADVC001 toward registrational development, expanding isotope supply and manufacturing infrastructure to support Phase 3 and future commercial demand, and progressing our Lead-212 pipeline into the clinic. Together, these priorities establish a clear path towards a diversified clinical pipeline with the goal of bringing the promise of targeted alpha therapy to more patients with cancer. ”
“The companies poised to lead the next generation of targeted alpha therapies will be those that combine differentiated clinical assets with end-to-end control over supply and manufacturing,” said Andrew Lam, PharmD, Managing Director, Head of Biotech Private Equity at Ally Bridge Group. “AdvanCell has assembled that foundation through its de-risked lead program, vertically integrated platform and experienced leadership team, uniquely positioning the company to execute at scale and emerge as a potential category leader.”
“The most enduring healthcare companies combine breakthrough science with the infrastructure and expertise to repeatedly develop new medicines,” said Nik Economopoulos, Director, Life Sciences Investments, Alpha Wave. “We believe AdvanCell is building that kind of generational company, with the platform, manufacturing capabilities and pipeline to unlock the full potential of targeted alpha therapies.”
Concurrent with the financing round, Andrew Lam of Ally Bridge Group and Nik Economopoulos of Alpha Wave will join AdvanCell’s Board of Directors.
AdvanCell's lead program, ADVC001, is an investigational Lead-212 PSMA-targeted alpha therapy for metastatic prostate cancer currently in Phase 2 clinical development (NCT05720130). Designed to selectively deliver potent alpha radiation to tumor cells while minimizing radiation exposure to healthy tissue, ADVC001 has the potential to address key limitations of PSMA radioligand therapy, including treatment resistance, tolerability challenges and the need for dose optimization. ADVC001 has demonstrated encouraging Phase 1b anti-tumor activity and favorable tolerability in patients with prostate cancer. These results support the continued advancement of ADVC001 while validating AdvanCell’s Lead-212 platform and its broader pipeline of next-generation targeted alpha therapies across additional cancer indications.
XXX
About AdvanCell
AdvanCell is a clinical-stage radiopharmaceutical company developing next-generation targeted alpha therapies for cancer. Through its vertically integrated platform spanning proprietary Lead- 212 technology, secure isotope supply, advanced manufacturing and clinical development, the Company is unlocking the full potential of targeted alpha therapy. With integrated operations across North America and Australia, AdvanCell is advancing a pipeline of differentiated precision radiopharmaceuticals designed to improve outcomes for patients with cancer worldwide. For more information, visit www.advancell.com.au and follow us on LinkedIn.
About Ally Bridge Group
Ally Bridge Group is a global healthcare investment firm focused on private and public high- impact life science innovation. Founded in 2013 by Frank Yu, the firm has led or co-led over $8 billion in healthcare transactions. The firm’s mission is to generate superior risk-adjusted returns for investors guided by the core principle of selective investment in healthcare innovation that addresses unmet medical needs. Ally Bridge Group has offices in New York and Hong Kong. For more information, visit https://ally-bridge.com/ or follow us on LinkedIn.
About Alpha Wave
Alpha Wave is a global alternative asset manager that has four main verticals: private equity, private credit, public markets, and insurance/retirement solutions. It is led by Rick Gerson, Navroz Udwadia, and Ryan Khoury. In private equity, Alpha Wave's objective is to invest in best- in-class growth stage companies defining category leadership in the AI era along with life science companies pursuing breakthrough innovations; in private credit, direct lending to established businesses seeking funding solutions; and in public markets an uncorrelated strategy. Alpha Wave is building an AI-native life insurance and retirement solutions company. Alpha Wave has offices in Miami, New York, London, Monaco, Madrid, Abu Dhabi, Tel Aviv, Bangalore, Mumbai, New Delhi, and Sydney. Alpha Wave’s investments include SpaceX, Anthropic, OpenAI, Cerebras, TikTok, Aman Resorts, Long Lake, Cognition, and HistoSonics.
Company Contact
contact@advancell.com.au
Media Contact
Kimberly Ha
KKH Advisors
917-291-5744
kimberly.ha@kkhadvisors.com

After leading Muna’s evolution into a clinical-stage company, Rita Balice-Gordon, Ph.D., will step down and join the Board of Directors; former iTeos President & CEO Michel Detheux, Ph.D., appointed Chief Executive Officer
Company remains focused on developing new therapies to treat neurodegenerative diseases like Alzheimer's, Parkinson’s, and ALS, with lead TREM2 agonist on track to enter Phase 2 studies in early Alzheimer’s in early 2027
COPENHAGEN, Denmark & BOSTON, June 30, 2026 – Muna Therapeutics (Muna), a biotechnology company pioneering novel therapeutics for neurodegenerative diseases, today announced a CEO transition. Rita Balice-Gordon, Ph.D., Muna’s founding CSO and CEO since 2021, will step down and join the Company’s Board of Directors. Michel Detheux, Ph.D., a seasoned biotech entrepreneur and former President and CEO of iTeos Therapeutics (NASDAQ: ITOS), has been appointed CEO, effective June 29, 2026.
Under Balice-Gordon’s leadership, Muna has evolved into a clinical-stage company with a differentiated discovery platform and dynamic pipeline focused on neurodegenerative diseases. Muna’s lead asset, MNA-001, is a best-in-class small-molecule TREM2 agonist that is completing Phase 1 and is on track to begin Phase 2 testing in early Alzheimer’s patients in early 2027. As the company continues to progress, Detheux's experience leading biotechs, advancing innovative therapies, and forging strategic alliances will help build on this momentum.
“It has been one of the greatest privileges of my career to build Muna and lead such a talented team dedicated to bringing hope to patients living with neurodegenerative disease,” said Balice-Gordon. “I am extraordinarily proud of what we have accomplished together: advancing MNA-001 toward Phase 2 clinical development; establishing our innovative MIND-MAP discovery platform grounded in human biology; and building a world-class team. I look forward to continuing to support Muna as a member of the Board and am excited to see the company continue to grow under Michel's leadership.”
Detheux brings more than 30 years of biotech leadership experience to this role. As the founder and President & CEO of iTeos, he led the company from a research spin-off to a publicly traded clinical-stage entity, raising more than $1.2B and expanding to 180 employees across the US and Europe. Under his leadership, the company also advanced multiple development programs and established significant strategic partnerships, including a $2B collaboration with GSK.
“Rita and the team have built a platform and pipeline unlike anything else in neurodegeneration," said Michel Detheux, Ph.D., incoming Chief Executive Officer. “Their exceptional work has yielded a compelling clinical asset in the TREM2 agonist program and a major validation of the MiND-MAP platform through the GSK alliance, a partnership model I know well. I am honored to take the helm at this moment and work alongside the team to advance Muna's promising pipeline for people living with neurodegenerative diseases.”
“On behalf of the entire Board, I want to express our profound gratitude to Rita for her leadership in shaping Muna into the company it is today,” said Donald Nicholson, Ph.D., Chair of Muna’s Board of Directors. “Her scientific vision, drive, and deep commitment to patients have been foundational to what we have achieved. We are delighted that she will continue to contribute her unique perspective as a Board member and extremely pleased to welcome Michel, whose business experience and acumen will be instrumental in guiding Muna through future clinical and corporate milestones.”
The announcement comes as Muna pushes its small molecule TREM2 agonist program to Phase 2 clinical evaluation and prepares for continued progress across its pipeline of novel neurodegeneration targets. In June, the company published a study in Nature Medicine, in which it leveraged its MiND-MAP platform to identify a critical tipping point in microglial response to amyloid and tau pathology, pinpointing microglial state transitions as key drivers of Alzheimer’s.
About Muna Therapeutics
Muna Therapeutics discovers and develops therapies that slow or stop devastating neurodegenerative diseases including Alzheimer’s and Parkinson’s disease. These disorders impact memory, movement, language, behavior and personality, resulting in disability and death of millions of patients around the globe. Muna focuses its groundbreaking science on identifying new medicines to preserve cognition and other brain functions, enhance resilience to disease pathology, and slow or stop the progression of neurodegenerative diseases. Its name reflects this focus: Muna means ‘to remember’ in Old Norse. Muna is headquartered in Copenhagen, Denmark, and has operations in the United States. For more information, visit www.munatherapeutics.com.
Follow Muna on LinkedIn.
Media Contact:
Lia Dangelico
Deerfield Group
lia.dangelico@deerfieldgroup.com
- New 128,000-square-foot facility in Andover will serve as U.S. global headquarters and future manufacturing center supporting the development and commercialization of ADVC001 and the Company’s pipeline of Lead-212 targeted alpha therapies
- Expanding U.S. operations complement the Company’s established Australian R&D and manufacturing capabilities to create a global, vertically integrated platform for radiopharmaceutical development and commercial supply
Boston, USA and Brisbane, Australia – June 22, 2026 – AdvanCell, a clinical-stage radiopharmaceutical company developing innovative targeted alpha therapies for cancer, today announced the establishment of its U.S. Global Headquarters in the Greater Boston area, and the lease of a new 128,000-square-foot facility that will become the Company’s U.S. flagship manufacturing center supporting its long-term growth and commercialization strategy. The expansion marks an important milestone in the Company’s evolution into a U.S. -based radiopharmaceutical company with integrated operations spanning North America and Australia.
The investment reflects AdvanCell's strategy to expand its U.S. presence and build the infrastructure needed to support the development and future commercialization of ADVC001 and its Lead-212 targeted alpha therapy pipeline.
Located in Greater Boston, the Innovation Park campus owned by IQHQ provides access to a world-class life-science ecosystem, supporting the next phase of the Company’s growth. The Andover facility will serve as the Company’s U.S. Global Headquarters, bringing together corporate operations and state-of-the-art manufacturing capabilities.
“Establishing AdvanCell’s U.S. Global Headquarters and future manufacturing facility reinforces our commitment to U.S. expansion and represents an important milestone in AdvanCell’s strategy to build a global, vertically integrated targeted alpha therapy company,” said Philina Lee, PhD, CEO of AdvanCell. “As we advance ADVC001 and our broader Lead-212 pipeline, our Andover facility will be a cornerstone of our U.S. expansion and our first internal manufacturing site in the U.S., providing the foundation to scale production to support future clinical and commercial demand. We will continue to leverage the strengths of our Australian operations - including rapid clinical translation, isotope supply capabilities and process innovation - as we advance our Lead- 212 targeted alpha therapies for patients globally.”
As part of its U.S. manufacturing expansion strategy, AdvanCell is also working with a leading contract development and manufacturing organization (CDMO) to establish drug product manufacturing capabilities in addition to the Andover facility fit out and qualification. This approach is expected to accelerate access to U.S.-based manufacturing capacity, supporting Phase 2 enrolment of the TheraPb study (NCT05720130) in the U.S. while the Company further builds its Phase 3 and commercial manufacturing infrastructure.
XXX
About 212Pb-ADVC001
212Pb-ADVC001 (ADVC001) is a proprietary and patented PSMA-targeting radioligand with optimized physicochemical properties and labelled with Lead-212 (212Pb), an alpha-emitting payload (radionuclide) with a high dose rate, 10.6-hour half-life and simple decay scheme. ADVC001 is designed to deliver radiation at a cellular level to effectively kill prostate cancer cells while minimizing toxicity.
About the TheraPb trial
The TheraPb trial (NCT05720130) is a prospective, open-label Phase 1/2 dose escalation and expansion study evaluating ADVC001 in metastatic prostate cancer. The completed Phase 1b dose escalation assessed the safety and tolerability of escalating doses of ADVC001 administered every 6, 4, 2 or 1 week(s) (see press release). The Phase 2 expansion is assessing the efficacy and safety of ADVC001 at two dose levels. The trial utilizes a randomized dose- response design and dose optimization elements to evaluate ADVC001 in PSMA-positive mCRPC and in mHSPC.
About AdvanCell
AdvanCell is a vertically integrated, clinical-stage radiopharmaceutical company dedicated to developing innovative cancer therapies that harness the power of targeted alpha-emitting radionuclides. By leveraging its proprietary Lead-212 platform, advanced and scalable manufacturing and world-class clinical development capabilities, AdvanCell aims to deliver novel treatments that improve outcomes for patients with cancer globally. For more information, visit www.advancell.com.au and follow us on LinkedIn.
Contacts
contact@advancell.com.au
For media inquiries, please contact:
MEDiSTRAVA (in the UK)
advancell@medistrava.com
+44 (0)20 3928 6700
- Series A co-led by Jeito Capital and Sofinnova Partners, with participation from Arkin Bio, Sanofi Ventures, Sixty Degree Capital, Vives Partners and Apollo Health Ventures
- Sofinnova Partners seed financed and co-founded Bionyra alongside Frédéric Marrache, MD, PhD, CEO of the company
- Pipeline includes clinical and IND stage mono and multispecific antibodies with half-life extension engineering across immune-mediated inflammatory diseases
PARIS and BOSTON, Mass. – June 22, 2026 – Bionyra Pharma, a clinical-stage biopharmaceutical company developing next-generation biologics for immune-mediated inflammatory diseases, today announced the company’s launch and its $165 million (~€143 million) oversubscribed Series A financing co-led by Jeito Capital and Sofinnova Partners. Arkin Bio, Sanofi Ventures, Sixty Degree Capital, Vives Partners and Apollo Health Ventures also participated in this inaugural funding. The company received seed financing from Sofinnova Partners and was co-founded with former Sanofi R&D executive Dr. Frédéric Marrache, who serves as CEO of the company. Bionyra is advancing next-generation mono and multispecific antibodies for diseases with high unmet need despite currently available treatments, including atopic dermatitis and inflammatory bowel disease (IBD).
“More than half of patients suffering from immune-mediated inflammatory diseases remain inadequately controlled, highlighting a significant need for better treatment options. Across our pipeline, we are selectively modulating immune pathways that are central drivers of disease in indications where current therapies remain insufficient,” said Dr. Marrache, Co-founder and CEO of Bionyra. “We have built Bionyra around assets and targets with clear therapeutic rationale, relevant biology and optimized clinical strategy to develop therapies that will deliver meaningful impact to people who don’t currently have good options.”
Severe immunological and inflammatory diseases impose a constant toll, as patients live with a high daily burden of pain, fatigue and uncertainty, structuring their lives around flares, hospital visits and treatment setbacks. Current therapies often target single nodes of complex immune networks, limiting depth and durability of response. Many patients cycle through multiple therapies without achieving sustained relief or meaningful improvements in quality of life, highlighting the need for better therapeutic options that can improve clinical outcomes.
Bionyra is launching with a broad pipeline of clinical and near-clinical, next-generation anti-inflammatory therapies designed to deliver first- and best-in-class treatments by combining multi-pronged biology with optimized pharmacokinetics to maximize efficacy and patient benefit, including:
- BYN-002 — a leading TL1A monoclonal antibody with half-life extension (HLE) technology with best-in-class potential in IBD and other TL1A-relevant indications. A Phase 1 study in healthy subjects is fully enrolled.
- BYN-003 — a first and best-in-class TL1A*IL-23p19 bispecific antibody with HLE technology that entered the clinic in April with a Phase 1 study now underway.
- BYN-001 — a first and best-in-class IL-25 monoclonal antibody with HLE technology in IND-stage for atopic dermatitis and Type 2 inflammation.
The company licensed BYN-002 and BYN-003 from TrueLab Biopharmaceutical1 and BYN-001 from NovaRock Biotherapeutics2 and holds exclusive global rights outside of Greater China. In addition, Bionyra is progressing additional innovative preclinical assets, including some from TrueLab.
“So many patients suffering from immune-mediated inflammatory diseases still face daily burden due to limited treatment options,” said Mehdi Ainouche, Partner at Jeito Capital. “Bionyra is uniquely positioned to accelerate delivery of highly promising therapies to patients through a focused and well-designed clinical development strategy aimed at rapidly generating clinical validation and addressing key unmet needs like deeper remission and alternatives for non-responders. We look forward to supporting Frédéric and the entire Bionyra team as the company reaches a pivotal stage in its development while simultaneously building a scalable pipeline in dermatology and IBD.”
“When we co-founded Bionyra with Frédéric, our conviction in both the company and his leadership was grounded in his deep expertise in immune and inflammatory diseases. Today, that conviction is reflected in a $165 million oversubscribed round backed by an exceptional syndicate of investors who share our belief in this pipeline and the team behind it. Looking ahead, we are focused on advancing these programs with the aim of bringing meaningful new treatment options to patients,” said Anta Gkelou, Partner at Sofinnova Partners.
Bionyra’s world-class team is led by Frédéric Marrache, MD, PhD, a former senior pharmaceutical executive with extensive experience in immunology and inflammation. Prior to joining Bionyra as CEO, Frédéric served as Vice President and Executive Global Project Head for Clinical Development in the Immunology and Inflammation Therapeutic Area at Sanofi, where he led a portfolio of early- to mid-stage drug development programs focused on immune-mediated diseases. Frédéric holds an MD in gastroenterology, a PhD, and an MBA.
Mehdi Ainouche, Partner at Jeito Capital; Anta Gkelou, Partner at Sofinnova Partners; Avital Adler, Principal at Arkin Bio; and Laia Crespo, Partner at Sanofi Ventures, will join Bionyra’s Board of Directors.
About Bionyra Pharma
Bionyra Pharma is a clinical-stage biopharmaceutical company developing nextgeneration biologics for severe immunological and inflammatory diseases. Bionyra’s best-in-class mono and multispecific antibodies target novel immune pathways, enabling deeper and more durable disease control, with an initial focus on atopic dermatitis and inflammatory bowel disease. Seed financed by Sofinnova Partners, Bionyra was co-founded by Sofinnova Partners and Frédéric Marrache, MD, PhD, MBA, a former Sanofi R&D executive with deep immunology expertise. The company is committed to overcoming the limitations of existing therapies by delivering patientoptimized treatments with greater efficacy and improved convenience, offering better and longer-lasting outcomes. For more information, visit Bionyra.com and follow us on LinkedIn.
About Sofinnova Partners
Sofinnova Partners is a leading European venture capital firm in life sciences, specializing in healthcare and sustainability. Based in Paris, London and Milan, the firm brings together a team of professionals from all over the world with strong scientific, medical and business expertise. Sofinnova Partners is a hands-on company builder across the entire value chain of life sciences investments, from seed to laterstage.
Founded in 1972, Sofinnova Partners is a deeply established venture capital firm in Europe, with 50 years of experience backing over 500 companies and creating market leaders around the globe. Today, Sofinnova Partners manages over €4 billion in assets. For more information, please visit: sofinnovapartners.com.
About Jeito Capital
Jeito Capital is a global leading private equity firm with a patient benefit driven approach that finances and accelerates the development and growth of ground-breaking medical innovation. With €1.6 billion in assets under management, Jeito empowers and supports managers through its expert, integrated, multi-talented team and through the investment of significant capital to ensure the growth of companies, building market leaders in their respective therapeutic areas with accelerated patients’ access globally, especially in Europe and the United States. Jeito has built a diversified portfolio of clinical biopharmas with cutting-edge innovations addressing high unmet needs. Jeito Capital is based in Paris with a presence in Europe and the United States. For more information, please visit www.jeito.life or follow us on LinkedIn.
About Arkin Bio Capital
Arkin Bio Capital is a global biotech fund dedicated to supporting clinical stage biotech companies approaching proof of concept in patients. Arkin Bio Capital leads investments alongside top global partners, driving robust growth and impactful advancements. Arkin’s team has a strong background in biopharmaceuticals, drug development, business development, and management, with a proven track record. Arkin Bio Capital seeks to invest in companies with innovative therapeutic candidates and experienced managements and is committed to nurturing promising opportunities and ensuring the success of each investment. For more information about Arkin Bio Capital, please visit https://arkin-capital.com/bio/.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi, focused on investing in promising early-stage healthcare companies. The firm supports pioneering innovations in biotechnology, digital health, and life sciences aligning with Sanofi’s mission to bring life-changing treatments to patients worldwide. For more information, please visit www.sanofiventures.com.
Media Contact
Michelle Linn
Linnden Communications
michelle@linndencom.com
1 BYN-002 is also known as TL-001 and BYN-003 as TL-003.
2 BYN-001 is also known as NBL-023.


- Lead program, MMT-205, licensed from MabTics Co., Ltd / Curacle Co., Ltd. is a potenNal best- in-class bispecific anNbody which acNvates Tie2 and inhibits VEGF, in development for neovascular age-related macular degeneraNon (nAMD) and diabeNc macular edema (DME)
- In connecNon with the transacNon, Memento Medicines has closed a $93M Series A financing, co-led by Forbion, RA Capital Management and Avego BioScience Capital, to support the progress of MMT-205 through IND-enabling and clinical studies
- Memento Medicines is the fiYh subsidiary financed by Sera Medicines, an anNbody accelerator formed in collaboraNon with RA Capital in 2023
Boston, MA – Memento Medicines, Inc., a privately-held biotech company developing biologic therapies for retinal diseases, today announced the completion of a $93M Series A financing round co-led by Forbion, RA Capital Management and Avego BioScience Capital, with participation from Sanofi Ventures and Samsara BioCapital. In connection with the financing, Memento also announced that it has entered into an exclusive license agreement with MabTics Co., Ltd. and Curacle Co., Ltd. to obtain worldwide rights to MMT-205. MMT-205, previously known as MT-103, is a bispecific antibody that activates Tie2 and inhibits VEGF , two clinically validated targets that play a central role in the pathogenesis of retinal and vascular diseases. Under the terms of the license agreement, MabTics and Curacle have received upfront consideration comprising cash payments and equity in Memento and are also eligible for additional development, regulatory, and commercial milestones and tiered royalties on net sales.
MMT-205’s agonism of Tie2 simultaneously with inhibition of VEGF may improve upon the established efficacy of anti-VEGF monotherapies in the treatment of retinal diseases such as neovascular age-related macular degeneration (nAMD) and diabetic macular edema (DME). MMT-205 activates Tie2 signaling through direct engagement with the Tie2 receptor . Compared to current therapies, MMT-205 produces greater Tie2 activation and improved integrity of the retinal vasculature in preclinical models. Proceeds from the financing will support IND-enabling studies for MMT-205, with clinical trials expected to initiate in 2027.
“MMT-205 holds the potential to become a best-in-class biologic therapy in nAMD and DME and is supported by a robust preclinical data package established by our partners at MabTics and Curacle,” said Naveen Daryani, CEO of Memento Medicines. “Memento is fortunate to have the backing of an accomplished investor syndicate with deep domain expertise in ophthalmology, enabling us to rapidly bring this therapy to patients with various retinal diseases.”
Dr. Dmitrij Hristodorov, General Partner at Forbion commented “Given Forbion’s experience in building and guiding ophthalmology companies, partnering with Memento allows MMT-205 to reach its full potential. We believe that MMT-205 has the potential to be a transformative therapeutic in retinal diseases. ”
Dr. Nam-Kyung Lee, CEO of MabTics, and Jaehyeon Ryu, CEO of Curacle remarked: “We are profoundly excited and proud to enter into this alliance, which reflects recognition of the strong therapeutic potential of MMT-205—a novel bispecific antibody that activates Tie2 and inhibits VEGF—compared to current clinical competitors. By leveraging Memento’s outstanding development capabilities and execution strength, we can project our scientific innovations globally. Together, we will actively collaborate to accelerate the clinical advancement of MMT-205, bringing impachul clinical solutions to patients with ocular diseases at an unprecedented pace.”
In conjunction with the financing, Dmitrij Hristodorov, Ph.D., General Partner at Forbion; Zach Scheiner, Ph.D., Partner at RA Capital; Vishal Kapoor, MBA, Partner at Avego BioScience Capital; and Megan Krench, Ph.D., Principal at Sanofi Ventures will join Memento Medicines’ Board of Directors. Memento Medicines was founded by RA Capital’s Sera Medicines, a biologics-focused accelerator advancing next- generation protein therapeutics.
About Memento Medicines
Memento Medicines is a biotech company advancing novel antibody therapies for ophthalmology. Founded in 2026 as the filh subsidiary of Sera Medicines, a biologics-focused antibody accelerator formed in collaboration with RA Capital, the Company is committed to developing new therapies for patients living with retinal diseases. Memento's lead program, MMT-205, is a potential best-in-class bispecific antibody that activates Tie2 and inhibits VEGF, in development for neovascular age-related macular degeneration (nAMD) and diabetic macular edema (DME).
About Forbion
Forbion is a leading global venture capital firm with deep roots in Europe and offices in Naarden, the Netherlands, Munich, Germany, and Boston, USA. Forbion invests in innovative biotech companies, managing approximately €5 billion across multiple fund strategies covering all stages of (bio)pharmaceutical drug development. In addition to its human health focus, Forbion also invests in planetary health solutions through its BioEconomy strategy. The firm’s team of over 30 investment professionals has a strong track record, with more than 130 investments across 11 funds, resulting in numerous approved therapies and successful exits. Forbion is a signatory to the UN Principles for Responsible Investment and operates a joint venture with BGV for seed and early-stage investments in Europe.
About RA Capital Management
Founded in 2004, RA Capital Management is a multi-stage investment manager dedicated to evidence- based investing in public and private healthcare, life sciences, and planetary health companies. RA Capital creates and funds innovative companies, from private seed rounds to public follow-on financings, allowing management teams to drive value creation from inception through commercialization and beyond. RA Capital's knowledge engine is guided by its dedicated, science-first internal research division and Raven, RA Capital's healthcare incubator, offers entrepreneurs and innovators a collaborative and comprehensive platform to explore the novel and the re-imagined. RA Capital has more than 200 employees and over $14 billion in assets under management.
About Avego BioScience Capital
Avego BioScience Capital is a mid-to-late stage life sciences venture firm led by an experienced team of investors and operators with over 100 years of combined biopharma experience. The firm employs a fundamentals-driven, asset-centric investment strategy focused on therapeutics companies. Avego BioScience Capital is currently investing from its second venture fund.
About MabTics
Founded in 2022 by experts with more than 15 years of experience in antibody engineering and translational research, MabTics is dedicated to delivering new hope to patients suffering from intractable vascular diseases with high unmet medical needs through the development of innovative antibody therapeutics based on its proprietary human antibody library with world-class diversity. Through the MMT-205/MT-103 licensing agreement with Memento Medicines, MabTics has further validated its capabilities in antibody drug discovery and development. The company will continue to advance a pipeline of differentiated antibody therapeutics, including vascular normalization-inducing antibodies for renal diseases and anti-thrombotic antibodies designed to address thrombotic disorders without bleeding liabilities.
About Curacle
Curacle Co., Ltd. (KOSDAQ: 365270) is a clinical-stage biopharmaceutical company focused on developing therapies for endothelial and microvascular dysfunction, key underlying pathologies associated with aging and chronic diseases. The company is advancing a pipeline of novel small-molecule therapeutics for retinal, renal, neurodegenerative, and vascular diseases, including Rivasterat (CU06), currently in Phase 2 development for diabetic macular edema (DME), and CU01 for renal diseases. Curacle is also collaborating with MabTics on the development of novel biologics targeting vascular dysfunction. Through its differentiated approach to vascular biology and collaborative biologics research, Curacle aims to develop innovative therapies for patients with serious diseases and high unmet medical needs.
Media and Investor Contact:
Lauren McCormick
ICR Healthcare
Lauren.McCormick@icrhealthcare.com
Data showed LFD-200 selectively delivers glucocorticoid payload to immune cells, resulting in dose- responsive anti-inflammatory activity without impact on cortisol - a sensitive measure of GC toxicity
Single doses ≤12 mg/kg and four weekly 5.5 mg/kg doses were safe and well tolerated in healthy participants
Short duration of exposure of LFD-200 in blood was consistent with rapid uptake into immune cells and tissue residency of ≥2 weeks, supporting potential for less frequent dosing
Dosing in the Phase 1 trial in patients with rheumatoid arthritis is ongoing
BURLINGTON, MA – June 8, 2026 -- Lifordi Immunotherapeutics, Inc., a clinical-stage biotechnology company developing antibody-drug conjugates (ADCs) for the treatment of autoimmune and inflammatory disorders, presented first-in-human data for LFD-200, the Company’s novel subcutaneously (SC) administered ADC delivering a potent glucocorticoid (GC) directly to immune cells. Phase 1 data from healthy participants (HPs) presented at EULAR 2026 (European Congress of Rheumatology), in London, UK, June 3-6, 2026, showed LFD- 200 was well tolerated and demonstrated dose-responsive anti-inflammatory activity with no impact on serum cortisol levels, a sensitive marker for systemic GC toxicity. Dosing patients with moderate to severe rheumatoid arthritis (RA) in the Phase 1 study is ongoing with data expected by year-end 2026.
“We believe these data provide proof of mechanism for our VISTA-targeted ADC approach, which is designed to deliver a therapeutic payload directly to immune cells while avoiding the side effects associated with systemic exposure,” said Arthur Tzianabos, Ph.D., President & Chief Executive Officer. “By leveraging the unique properties of LFD-200 — including rapid uptake into immune cells, prolonged tissue residency, and anti-inflammatory activity — we believe we are establishing a differentiated approach to the treatment of autoimmune and inflammatory diseases. We look forward to seeing LFD-200 data in patients with RA.”
LFD-200 is a novel ADC composed of a monoclonal antibody (mAb) that is conjugated to 8 glucocorticoid payloads. The mAb is designed to selectively bind to the immune cell surface protein V-domain immunoglobulin suppressor of T cell activation (VISTA), rapidly internalize the GC payload, and have no other biological function. Nonclinical studies of subcutaneous dosing with LFD-200 in non-human primates1 (NHP) demonstrated sustained GC payload exposures in lymph nodes and spleen. Additionally, ex vivo stimulation of proinflammatory cytokines in whole blood and bone marrow samples was suppressed in a dose- responsive manner and no impact on cortisol was seen in any study, including after 13 weekly doses.
The Phase 1 clinical trial is a single and multiple ascending dose (SAD/MAD), randomized, double-blind, placebo- and active-controlled first-in-human study designed to evaluate the safety, efficacy, pharmacokinetics (PK) and pharmacodynamics (PD) of LFD-200 in healthy participants and patients with moderate to severe RA.
Part 1: HPs [N=11/cohort (SAD) or N=8/cohort (MAD)]
- SAD HP were randomized to receive a single dose of either oral prednisone (10 mg, open label; N=3), placebo (N=2), or SC LFD-200 (N=6; 1.5, 3, 6, or 12 mg/kg dose)
- MAD HP were randomized to receive 4 weekly 5.5 mg/kg doses of SC LFD-200 (N=6) or placebo (N=2)
Part 2: Cohorts of patients with moderate to severe RA (N=14/cohort)
- Randomization in each cohort is 8 LFD-200: 3 placebo: 3 prednisone with dosing for 12 weeks
In the poster (POS#1310) entitled “LFD-200, an Antibody Drug Conjugate that Selectively Delivers a Glucocorticoid Payload to Immune Cells, has a Favorable Risk-Benefit Profile vs. Prednisone and Placebo in a First in Human Study,” presented on Saturday, June 6, 2026, [10:15 am local time], Dr. Matthew W. McClure, Chief Medical Officer of Lifordi, shared data from 4 SAD and 1 MAD cohorts of HPs, which demonstrated:
- LFD-200 was safe and well tolerated
- No serious adverse events
- Treatment-emergent adverse events: No concerning findings/trends (Grade ≤2)
- Laboratories/vital signs/electrocardiograms/physical examination: no concerning findings or trends
- LFD-200 plasma exposures were supportive of biweekly or monthly dosing
- Serum half-life of ADC was ~30 hours, which is consistent with targeted uptake into VISTA+ cells
- Steady state reached after 2nd dose
- Free GC payload exposure persists beyond ADC detection, suggesting tissue exposure ≥2 weeks
- LFD-200 had no impact on cortisol levels, a sensitive measure of systemic GC toxicity, at any time point after doses as high as 12 mg/kg (equivalent to ~170 mg of prednisone in a 70 kg HP)
- LFD-200 showed sustained and potent GC activity in whole blood*
- TNFα levels after ex vivo lipopolysaccharide stimulation of whole blood samples in the SAD/MAD study demonstrated a dose-responsive effect for LFD-200 that was comparable to a single 10 mg dose of prednisone and was sustained. A similar effect was seen for IL-6 and IL-1β.
*Blood is not target tissue; Assay known to underrepresent in vivo effects in immune tissues
“These results demonstrate successful translation of our preclinical LFD-200 findings into humans and further validate our ability to deliver a potent glucocorticoid to immune cells and avoid the systemic side effects that have limited their use for decades,” said Dr. McClure. “Importantly, the safety profile observed in healthy participants — including no impact on cortisol levels — together with reductions in proinflammatory cytokines, strengthens our confidence that these findings may translate into meaningful benefit for patients with RA.”
For further details, view the poster here.
About Lifordi
Lifordi Immunotherapeutics, Inc. is a clinical-stage biotechnology company leading the way by leveraging the success of antibody-drug conjugates (ADCs) to develop treatments for autoimmune and inflammatory disorders. The Company’s lead ADC, LFD-200, is in a Phase 1 clinical trial for rheumatoid arthritis and demonstrated a favorable safety, PK and PD profile in healthy participants, including dose-responsive anti- inflammatory activity without systemic glucocorticoid toxicity (cortisol suppression). Lifordi has also applied its novel drug delivery approach to other diverse payloads, such as antisense oligonucleotides (ASOs), siRNA, and small molecules. As experienced drug developers in immunology and inflammatory diseases, together with expert clinical advisors and funding from ARCH Venture Partners, Atlas Venture, 5AM Ventures, and Sanofi Ventures, Lifordi is committed to changing how immune and inflammatory diseases are treated. For more information, please visit www.lifordi.com and follow us on LinkedIn.
Contact
Theresa McNeely
Chief Communications Officer
tmcneely@lifordi.com
(508) 523-9511
1McClure et al., ACR 2025
- Justyna Kelly, MSc, joins as Chief Technology Officer and François Gaudet, PhD, joins as Chief Scientific Officer, further building AdvanCell’s U.S. based leadership team
- Simon Puttick, PhD, transitions to Chief Isotope Development Officer and continues to lead isotope innovation efforts from Australia
- Appointments reflect AdvanCell’s continued investment in pipeline advancement, global scale-up, manufacturing readiness and isotope capabilities
Boston, USA and Brisbane, Australia – June 8, 2026 – AdvanCell, a clinical-stage radiopharmaceutical company developing innovative targeted alpha therapies for cancer, today announced the appointment of Justyna Kelly as Chief Technology Officer (CTO) and François Gaudet as Chief Scientific Officer (CSO), expanding the company’s executive leadership team in the U.S. as it continues to scale its U.S. capabilities, and advance its growing pipeline of innovative targeted alpha therapies.
Justyna Kelly will lead AdvanCell’s global technical operations strategy, including CMC, GMP production and clinical supply, the company’s global manufacturing network, supply chain, and preparation for commercial readiness. Her focus will include further developing U.S. manufacturing capabilities to support the next phase of development for ADVC001. Ms. Kelly brings 16 years of radiopharmaceutical manufacturing and operations experience, most recently serving as Vice President and Site Head of Eli Lilly and Company’s Indianapolis radioligand therapy manufacturing site. She previously served as Chief Operating Officer at POINT Biopharma prior to its acquisition by Lilly and has held leadership roles of increasing responsibility, including Chief Executive Officer, at the Centre for Probe Development and Commercialization. She holds an MSc in Biochemistry from McMaster University.
“AdvanCell has built a differentiated Lead-212 platform with the potential to meaningfully impact patients with cancer,” said Justyna Kelly, CTO of AdvanCell. “I am excited to join the team and help execute a holistic strategy to scale our Lead-212 manufacturing infrastructure for Phase 3 and commercial readiness, while ensuring the quality, reliability, and operational excellence needed to deliver these therapies to patients.”
François Gaudet has commenced the role of CSO and will lead AdvanCell’s discovery and preclinical efforts, with a focus on accelerating the progression of new innovative Lead-212 targeted alpha therapies into clinical development. An accomplished drug hunter, Dr. Gaudet brings 25 years of experience across pharmaceutical and biotechnology companies, including leadership roles of increasing responsibility at Novartis, Johnson & Johnson, and Mnemo Therapeutics where he served as Chief Scientific Officer. He has led research teams responsible for the discovery and development of innovative therapeutic assets through commercialization, including TECVAYLI® and TALVEY® . He conducted his graduate work in cancer epigenetics at the Massachusetts Institute of Technology and holds a PhD in Biology from Ludwig Maximilians Universität.
“AdvanCell has built an innovative platform of Lead-212 targeted alpha therapies based on its commitment to advance new treatment options for patients in oncology. Lead-212 alpha therapy is a nascent field and ADVC001 represents a validation of the modality,” said François Gaudet, CSO at AdvanCell. “I’m excited to have joined the team to continue driving forward the pipeline development and help realize the full potential of the company’s differentiated approach.”
Simon Puttick has transitioned from Chief Scientific Officer to the role of Chief Isotope Development Officer. Having played a key role in establishing AdvanCell’s targeted alpha therapy platform, Dr. Puttick, in his new role, will have a dedicated mandate to advance the company’s next-generation isotope production technologies, driving radioisotope process development from concept through to scalable manufacturing, strengthening supply chain security, and further positioning AdvanCell as a leader in this strategically critical capability, supporting the company’s platform expansion. Dr. Puttick will continue to be based at the company’s Australian headquarters in Brisbane.
“These leadership appointments strengthen AdvanCell’s ability to build a category-defining targeted alpha therapy company, accelerating our U.S. expansion and scaling our targeted alpha therapy platform globally,” said Philina Lee, CEO of AdvanCell. “Justyna brings deep radiopharmaceutical manufacturing and operations expertise, François brings a proven track record of discovering and advancing innovative oncology therapeutics, and Simon’s dedicated focus on innovative methods for next-generation isotope production reinforces a core strategic advantage for AdvanCell: secure, scalable isotope supply and manufacturing.”
TECVAYLI® and TALVEY® are registered trademarks of Johnson & Johnson or its affiliated companies.
XXX
About 212Pb-ADVC001
212Pb-ADVC001 (ADVC001) is a proprietary and patented PSMA-targeting radioligand with optimized physicochemical properties and labelled with Lead-212 (212Pb), an alpha-emitting payload (radionuclide) with a high dose rate, 10.6-hour half-life and simple decay scheme. ADVC001 is designed to deliver radiation at a cellular level to effectively kill prostate cancer cells while minimizing toxicity.
About the TheraPb trial
The TheraPb trial (NCT05720130) is a prospective, open-label Phase 1/2 dose escalation and expansion study evaluating ADVC001 in metastatic prostate cancer. The completed Phase 1b dose escalation assessed the safety and tolerability of escalating doses of ADVC001 administered every 6, 4, 2 or 1 week(s) (see press release). The Phase 2 expansion is assessing the efficacy and safety of ADVC001 at two dose levels. The trial utilizes a randomized dose- response design and dose optimization elements to evaluate ADVC001 in PSMA-positive mCRPC and in mHSPC.
About AdvanCell
AdvanCell is a vertically integrated, clinical-stage radiopharmaceutical company dedicated to developing innovative cancer therapies that harness the power of targeted alpha-emitting radionuclides. By leveraging its proprietary Lead-212 platform, advanced and scalable manufacturing and world-class clinical development capabilities, AdvanCell aims to deliver novel treatments that improve outcomes for patients with cancer globally. For more information, visit www.advancell.com.au and follow us on LinkedIn.
Contacts
contact@advancell.com.au
For media inquiries, please contact:
MEDiSTRAVA (in the UK)
advancell@medistrava.com
+44 (0)20 3928 6700

Proposed acquisition to add VGA039, a novel investigational monoclonal antibody that targets Protein S in Phase 3 development for von Willebrand disease (VWD)
Star Therapeutics to receive $1.25 billion upfront, with up to $750 million in additional payments upon achievement of sales milestones
Incyte will host an analyst and investor call on Monday, June 8, 2026, at 8:00 a.m. ET
WILMINGTON, Del.--(BUSINESS WIRE)--Jun. 8, 2026-- Incyte (Nasdaq:INCY) announced today it has entered into a definitive agreement to acquire Vega Therapeutics, Inc., a wholly owned subsidiary of Star Therapeutics, LLC, for $1.25 billion. Star Therapeutics will be eligible to receive up to $750 million in additional payments upon the achievement of sales milestones, for total potential consideration of up to $2.0 billion subject to customary closing adjustments. The proposed acquisition would add VGA039, a novel monoclonal antibody, to Incyte’s hematology portfolio.
Vega Therapeutics’ lead candidate, VGA039, modulates Protein S to improve hemostasis, potentially improving the body’s ability to control bleeding in numerous bleeding disorders. VGA039 is in Phase 3 pivotal development for patients with von Willebrand disease (VWD), the most common inherited bleeding disorder. It has the potential to be the first subcutaneous prophylactic therapy with a convenient dosing regimen for patients with VWD who currently require frequent intravenous infusions.
“VGA039 fits directly into our strategy of building a top-tier growth company for the future,” said Bill Meury, Chief Executive Officer of Incyte. “It is a first-in-class, Phase 3 asset with compelling early data, a manageable development path and the potential to become an important new growth driver in one of our core therapeutic areas – hematology. The transaction has all of the attributes we look for in business development opportunities.”
Approximately 135,000 people in the United States have been diagnosed with von Willebrand disease.1 The disease is characterized by excessive bleeding that can vary in severity and frequency and may significantly affect quality of life. Current prophylactic treatment options include factor replacement therapies that often require 2 to 3 intravenous infusions each week.2
“This milestone reflects our team’s deep commitment to innovation and underscores our strategy to develop first-in-class and best-in-class therapies for serious conditions with high unmet need,” said Adam Rosenthal, Ph.D., Founder and Chief Executive Officer of Star Therapeutics. “VGA039 will be advanced by Incyte, a global biopharmaceutical leader with deep expertise in hematology and a significant commercial track record. I am immensely proud of the Star Therapeutics team and our work toward making a difference for patients with von Willebrand disease.”
VGA039 has received Breakthrough Therapy, Fast Track, orphan drug and rare pediatric disease designations from the U.S. Food and Drug Administration (FDA). VGA039 has advanced into the Phase 3 VIVID-6 study (NCT07115004), a global single arm cross-over study to investigate safety and efficacy of the subcutaneous administration of VGA039 as prophylaxis for bleeding in patients with every type of VWD, including those with a high disease burden.
The transaction has been approved by both Incyte’s and Star Therapeutics’ Boards of Directors. Under the terms of the stock purchase agreement, Incyte will acquire all the outstanding shares of Vega Therapeutics, a wholly owned subsidiary of Star Therapeutics. The closing of the proposed transaction will be subject to certain conditions, including the expiration of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act and other customary conditions. The transaction is an equity acquisition and is expected to close in the third quarter of 2026, pending Hart-Scott-Rodino review resulting in an expected R&D charge of approximately $1.25 billion, that will be included in third quarter and full year 2026 GAAP and non-GAAP results.
Lazard is acting as financial advisor to Incyte and Goodwin Procter LLP is serving as its legal counsel. Evercore and Morgan Stanley are acting as financial advisors to Star Therapeutics, and Fenwick & West LLP is serving as its legal counsel.
Incyte Conference Call and Webcast
Incyte will host a conference call and webcast on Monday, June 8, 2026, at 8:00 a.m. ET to discuss the acquisition.
To access the conference call, please dial 877-407-3042 for domestic callers or 201-389-0864 for international callers. When prompted, provide the conference identification number, 13761011. If you are unable to participate, a replay of the conference call will be available for 30 days. The replay dial-in number for the United States is 877-660-6853 and the dial-in number for international callers is 201-612-7415. To access the replay, you will need the conference identification number, 13761011.
The live and archived webcast will be available via the Events and Presentations tab of the Investor section of Incyte.com.
About VGA039
VGA039 is an investigational monoclonal antibody therapy with a novel mechanism of action that targets Protein S, with dual actions promoting platelet attachment and enhancing fibrin deposition to restore hemostasis. VGA039 has the potential to be a universal hemostatic therapy that can treat numerous bleeding disorders, starting with all types of von Willebrand disease (VWD). As a subcutaneously self-administered investigational antibody therapy with a convenient once monthly dosing regimen, VGA039 has the potential to meaningfully improve convenience and quality of life for patients.
VGA039 has received Fast Track, orphan drug, rare pediatric disease and Breakthrough Therapy designations from the U.S. Food and Drug Administration (FDA). VGA039 has advanced into a Phase 3 study (NCT07115004), VIVID-6, a global single arm cross-over study designed to investigate the safety and efficacy of subcutaneous administration of VGA039 as prophylaxis for bleeding in patients with every type of VWD.
About von Willebrand Disease
Von Willebrand disease (VWD) is the most common inherited bleeding disorder in which the blood does not clot properly, caused by low or defective von Willebrand factor (VWF). VWD patients may experience excessive bleeding with varying severity and frequency, negatively impacting their daily lives. Current therapies for VWD prophylaxis include factor replacement therapies requiring multiple intravenous (IV) infusions every week. Approximately 135,000 people in the United States have been diagnosed with von Willebrand disease.1
About Star Therapeutics
Star Therapeutics is a biotechnology company focused on the discovery and development of life-changing therapies for diseases with significant unmet need. Star Therapeutics' team has invented four first-in-class antibody therapies, including the first approved drug (Enjaymo®) for cold agglutinin disease, and three other therapies that are each in Phase 3 development. For more information, please visit Star-Therapeutics.com and follow us on LinkedIn and X.1
About Incyte®
Incyte is redefining what’s possible in biopharmaceutical innovation. Through deep scientific expertise and a relentless focus on patients, we have built an established portfolio of first-in-class medicines and an extensive portfolio of next-generation medicines across our key franchises: Hematology, Oncology and Inflammation & Autoimmunity.
To learn more, visit Incyte.com and Investor.Incyte.com. Follow us on social media: LinkedIn, X and Instagram.
Incyte Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995 and other federal securities laws, including statements regarding the anticipated benefits of the Vega Therapeutics acquisition; costs and other anticipated financial impacts of the acquisition; expectations regarding VGA039’s development and its potential to become an important new growth driver for Incyte’s hematology portfolio; the potential and promise VGA039 offers patients with bleeding disorders and its ability to address significant unmet need; Incyte’s strategy of building a top-tier growth company for the future; expectations regarding the closing of the proposed transaction, including the expected timing of the same; and Incyte’s aspirations and goals as set forth under the heading “About Incyte.”
Actual results may differ materially from those indicated in the forward-looking statements as a result of various important factors, including unexpected costs, charges or expenses resulting from the acquisition; the risk that Incyte may not be able to successfully integrate the business of Vega Therapeutics and realize the expected benefits of the acquisition in a timely manner or at all; the sufficiency of clinical trial data for VGA039, as well as Incyte’s other products and product candidates, to meet applicable regulatory standards or warrant continued development; the ability to enroll sufficient numbers of subjects in clinical trials; actions of regulatory agencies, which may affect the initiation, timing and progress of clinical trials and marketing approval; Incyte’s ability to achieve commercial success for VGA039, if approved; Incyte’s ability to obtain and maintain protection of intellectual property for its products and technology; Incyte’s reliance on third parties and partners; the acceptance of Incyte’s products in the marketplace; market competition, sales, marketing, manufacturing and distribution requirements; and those risks and uncertainties discussed in greater detail in Incyte’s reports filed with the U.S. Securities and Exchange Commission, including its annual report on Form 10-K and its quarterly report on Form 10-Q for the quarter ended March 31, 2026. Incyte disclaims any intent or obligation to update these forward-looking statements.
Media
Argot Partners
media@incyte.com
Investors
ir@incyte.com
Source: Incyte
1Data on File.
2Franchini M, et al. Prophylactic management of patients with von Willebrand disease. Ther Adv Hematol. 2021;12.

Data published in the Journal of the American College of Cardiology Represents first peer-reviewed publication of clinical data demonstrating reductions in cardiometabolic risk markers with NLRP3 inhibitor
Simultaneous late-breaking presentation of these data at the 94th European Atherosclerosis Society Congress
NodThera is preparing to advance ruvonoflast into Phase 3 cardiometabolic development following the RESOLVE-1 trial readout expected in Q3 2026
Boston, MA, May 26, 2026 – NodThera, the leading clinical-stage NLRP3 company developing a
portfolio of potentially best-in-class brain-penetrant oral NLRP3 inhibitors to address inflammatory
drivers of cardiometabolic and neurologic diseases, today announced the publication of positive data
from its clinical trial evaluating the effects of ruvonoflast, previously known as NT-0796, in patients with cardiometabolic disease, in the Journal of the American College of Cardiology (JACC)1.
Concurrently, these data are being presented in a late-breaking oral session being held today at the 94th European Atherosclerosis Society (EAS) Congress in Athens, Greece.
“Inflammation is a causal, modifiable risk factor for atherosclerotic disease,” said Prof. Paul Ridker of the Harvard Medical School. “Our field now stands at an important inflection point, with multiple anti-inflammatory therapies in late-stage development. If also proven successful at reducing cardiovascular events, oral anti-inflammatory options that are scalable would be a welcome addition to our growing cardiovascular armamentarium.”
Data published and presented include the results from a randomized, double-blind, placebo-controlled
study to assess the effect of ruvonoflast on inflammation in participants at risk of cardiovascular disease (NCT06129409).
Highlights:
- Data provides evidence that upstream inhibition of the NLRP3/IL-1/IL-6/IL-18 inflammation pathway can achieve clinically significant reductions in validated biomarkers of cardiovascular risk.
- Ruvonoflast lowered hsCRP, a well-validated, independent, modifiable marker of inflammation associated with ASCVD events, by 82% in patients at high risk of ASCVD.
- 76% of ruvonoflast-treated patients achieved hsCRP <2 mg/L, the threshold shown to be associated with 25% MACE reduction in the landmark CANTOS cardiovascular outcome trial2.
- Ruvonoflast effects on hsCRP were rapid, sustained and readily reversible, a potentially key differentiator versus injectable biologics.
- Ruvonoflast significantly reduced key thrombo-inflammatory biomarkers beyond hsCRP, including IL-6, fibrinogen, and Lp(a). Taken together, these data underpin the antiinflammatory, anti-thrombotic and metabolic potential for ruvonoflast.
- Safety and efficacy data from this trial, as well as from NodThera’s previously published trial of patients with neuroinflammation3 informed the design of two large ongoing Phase 2 trials, RESOLVE-1 and RESOLVE-2, which are the largest and longest NLRP3 clinical trials to date.
“This study in patients with elevated residual inflammation establishes the potential of ruvonoflast as a selective anti-inflammatory therapy for people across the cardio-kidney-metabolic spectrum with elevated residual inflammation who remain at an increased risk for adverse cardiovascular outcomes,”
said Dr. Jyothis George, M.D., Ph.D., FRCP, FACE, Chief Medical Officer of NodThera. “We look forward to the upcoming readouts of both the RESOLVE-1 and RESOLVE-2 trials, and advancing ruvonoflast into Phase 3 development to serve patients living with cardiometabolic diseases.”
About the Journal of the American College of Cardiology (JACC)
JACC is the flagship peer-reviewed journal of the American College of Cardiology and is recognized as
one of the world’s leading cardiovascular journals. JACC publishes original clinical and translational
research, state-of-the-art reviews, and expert consensus documents focused on advancing the
prevention, diagnosis, and treatment of cardiovascular disease. Through rigorous scientific standards
and global reach, JACC serves cardiologists, researchers, and healthcare professionals worldwide.
About NodThera
NodThera, the leading clinical-stage NLRP3 company developing a portfolio of potentially best-in-class
brain-penetrant oral NLRP3 inhibitors to address inflammatory drivers of cardiometabolic and
neurologic diseases driven by the NLRP3/IL-1/IL-6/IL-18 inflammation pathway. The Company’s lead
program, ruvonoflast (NT-0796), is in Phase 2 cardiometabolic clinical trials which are expected to read
out in Q3 and Q4 2026, respectively. The Company expects to commence a Phase 3 registrational
study of ruvonoflast in H1 of 2027. The Company is also advancing NT-0150 for treatment of neuroinflammatory diseases and expects to release Phase 1 results in the H2 of 2026.
NodThera is backed by top-tier investors including Blue Owl Capital, Novo Holdings, F-Prime Capital,
5AM Ventures, Sofinnova Partners, Epidarex Capital, and Sanofi Ventures.
NodThera is headquartered in Boston, Massachusetts, with an R&D base in the UK.
Learn more at www.nodthera.com or follow the Company on LinkedIn.
Investors and Media
Argot Partners
nodthera@argotpartners.com
1 Ray KK, Clarke N, Thornton P, Miles AE, Digby Z, Davies MJ, Gorman M, Mullen B, Reader V, Magill M, Johnstone H, Ariti C,
Sattar N, Marx N, Navar AM, Hernandez AF, George JT, Watt AP, Butler J, Ridker PM. Anti-inflammatory effects of oral NLRP3
inhibition with ruvonoflast among individuals at elevated cardiovascular risk. JACC. 2026.
https://doi.org/10.1016/j.jacc.2026.05.014
2 Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ; CANTOS Trial Group. Relationship of C-reactive protein
reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS
randomised controlled trial. Lancet. 2018 Jan 27;391(10118):319-328. doi: 10.1016/S0140-6736(17)32814-3.
3 Clarke, N., Thornton, P., Reader, V., Lindsay, N., Digby, Z., Mullen, B., Gorman, M., Jacobson, E., Langdon, G., Johnstone, H., Wheeler, A. and Watt, A.P. (2025), Anti-Neuroinflammatory and Anti-Inflammatory Effects of the NLRP3 Inhibitor NT-0796 in Subjects with Parkinson's Disease. Mov Disord, 40: 2199-2208. https://doi.org/10.1002/mds.30307

Curevo’s lead asset, amezosvatein, is a Phase 3-ready shingles vaccine with potential to increase vaccination rates by delivering improved tolerability versus approved products
SEATTLE, May 26, 2026 (GLOBE NEWSWIRE) -- Curevo Vaccine (Curevo), a clinical-stage biotechnology company dedicated to developing varicella zoster virus (VZV) vaccines with improved tolerability, today announced entry into a definitive agreement to be acquired by Lilly.
Curevo’s lead product candidate is amezosvatein, an adjuvanted subunit vaccine for the prevention of shingles in adults. While the current standard of care for shingles prevention is effective, tolerability challenges can limit the overall vaccination rates and contribute to second-dose hesitancy, leaving a meaningful portion of patients with reduced or no protection against shingles and its long-term consequences. Amezosvatein was engineered with a next-generation synthetic adjuvant to overcome this problem. In a Phase 2 clinical trial head-to-head against the standard of care, amezosvatein matched immune response across all primary endpoints and reduced side effects such as activity-limiting fatigue, chills, and pain at the injection site by more than half. Given growing evidence linking shingles to elevated risk of stroke, and that shingles vaccination is associated with reduced dementia risk, a meaningfully better-tolerated vaccine could expand the reach of shingles prevention and reduce these long-term risks at a population level.
“Curevo is focused on improving the shingles immunization experience so more adults can benefit from protection against shingles, a serious disease with significant risk for long-term impairment of healthy living,” said George Simeon, chief executive officer of Curevo, Inc. “Lilly’s global development and commercialization capabilities will accelerate and expand upon amezosvatein's significant potential.”
“There is a growing body of evidence linking protection from shingles to lowered risk of stroke and dementia. A vaccine that is meaningfully better tolerated could extend the reach of shingles prevention and reduce these long-term risks at a population level,” said Daniel M. Skovronsky, M.D., Ph.D., Lilly's chief scientific and product officer, and president, Lilly Research Laboratories. “We look forward to working with the Curevo team to advance amezosvatein into Phase 3 and deliver a differentiated shingles vaccine to the millions of patients who remain unprotected."
Under the terms of the agreement, Lilly will acquire Curevo and Curevo shareholders could receive up to $1.5 billion in cash, inclusive of an upfront payment and a subsequent payment upon achievement of a specified milestone.
The transaction is subject to customary closing conditions, including expiration of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976.
Centerview Partners LLC and J.P. Morgan Securities LLC are acting as financial advisors and Cooley LLP is acting as legal advisor.
About Curevo Vaccine
Curevo is a privately held, clinical stage biotechnology company based near Seattle dedicated to reducing the burden of infectious disease by developing vaccines with improved tolerability and accessibility. Curevo’s lead product is amezosvatein, a non-mRNA adjuvanted sub-unit vaccine to prevent shingles, a serious medical condition involving a painful, blistering skin rash where 10-18% of people also develop serious, long-lasting nerve pain. For more information visit https://curevovaccine.com/.
Boston, MA, May 19, 2026 – NodThera, the leading clinical-stage NLRP3 company developing a portfolio of potentially best-in-class brain-penetrant oral NLRP3 inhibitors to address inflammatory drivers of cardiometabolic and neurologic diseases, today announced that data from the cardiometabolic Phase 1b clinical trial of its lead candidate, ruvonoflast, previously known as NT-0796, has been accepted for a late-breaking oral presentation at the 94th European Atherosclerosis Society (EAS) Congress being held May 24-27, 2026 in Athens, Greece.
Ruvonoflast is designed to be an oral, brain-penetrant NLRP3 inhibitor that reduces persistent inflammation that is a validated risk factor for cardiovascular events in patients living with cardiometabolic diseases.
“Many patients remain at high risk of cardiovascular events due to persistent inflammation,” said Professor Kausik K. Ray, Department of Primary Care and Public Health, Imperial College London. “The 2025 American College of Cardiology Scientific Statement emphasizes inflammation as a causal and clinically actionable driver of atherosclerotic cardiovascular diseases, independent of other drivers like LDL cholesterol. Targeting the upstream inflammatory node of NLRP3 represents a compelling therapeutic strategy to address this significant unmet need.”
The late-breaking presentation will highlight data on the anti-inflammatory effects of ruvonoflast in patients with cardiometabolic disease, including the effect on reduction in high-sensitivity C reactive protein (hsCRP) compared to injectable IL-6 therapies in development. In addition, the presentation will address the safety and tolerability of ruvonoflast in people with residual inflammatory risk.
“The European Atherosclerosis Society Congress is a premier global forum for advancing the understanding and treatment of cardiovascular disease,” said Jyothis George, M.D., Ph.D., FRCP, FACE, Chief Medical Officer of NodThera. “We are pleased to present the potential of ruvonoflast as a differentiated and pioneering therapeutic approach targeting inflammation in cardiometabolic disease. We are looking forward to upcoming data readout from RESOLVE-1, our Phase 2 study, in 3Q 2026. Compelling safety and efficacy data from these trials underpin ongoing planning to advance ruvonoflast into Phase 3.”
Details of the oral presentation are as follows:
Title: Oral NLRP3 inhibitor ruvonoflast provides rapid, robust and reversible inflammation reduction in people with residual inflammatory risk of ASCVD
Presenter: Prof. Kausik K. Ray, M.D., FMedSci
Abstract Number: 1629
Session Title: Late Breaker Clinical Abstracts
Date and Time: Tuesday, May 26, 2026, 15:45 - 16:00 EET
Location: Nanna Hall
About NodThera
NodThera is the leading clinical-stage NLRP3 company developing a portfolio of potentially best-in-class and first-in-class brain-penetrant and immune-targeted oral inhibitors to address unmet needs in cardiometabolic and neuroinflammatory diseases driven by the NLRP3/IL-1/IL-6/IL-18 inflammation pathway. The Company’s lead program, ruvonoflast (NT-0796), is in two Phase 2 cardiometabolic clinical trials which are expected to read out in Q3 and Q4 2026, respectively. The Company expects to commence a Phase 3 registrational study of ruvonoflast in H1 of 2027. The Company is also advancing NT-0150 for treatment of neuroinflammatory diseases and expects to release Phase 1 results in the H2 of 2026.
NodThera is backed by top-tier investors including Blue Owl Capital, Novo Holdings, F-Prime Capital, 5AM Ventures, Sofinnova Partners, Epidarex Capital, and Sanofi Ventures.
NodThera is headquartered in Boston, Massachusetts, with an R&D base in the UK.
Learn more at www.nodthera.com or follow the Company on LinkedIn.
Investors and Media
Argot Partners
nodthera@argotpartners.com

Partnering with leading pharmaceutical companies, BranchLab replaces fragmented, offline workflows with a unified, privacy-first AI platform—driving a nearly 70% increase in commercialization efficacy.
BOULDER, CO — [May 13, 2026] — BranchLab, an AI platform transforming how pharmaceutical companies commercialize therapies, today announced a $26 million Series A financing led by McKesson Ventures, with participation from FCA Venture Partners, Sanofi Ventures, and AIX Ventures. The round brings total funding to $35 million.
Pharma commercialization—spanning patient identification, audience segmentation, activation, and real-world measurement—has historically relied on fragmented vendors, delayed analytics, and manual workflows. The result is a system that is data-rich but operationally offline, limiting how quickly teams can learn, adapt, and drive impact.
BranchLab is changing that.
The company has built a unified AI platform that puts audience identification, activation, and optimization directly in the hands of pharma teams and their agencies—enabling them to act on high-intent patient and healthcare professional (HCP) audiences in near real time. Today, BranchLab is used by leading pharmaceutical companies and has delivered an average increase of nearly 70% in marketing efficacy across a diverse set of therapeutic areas.
“Pharma has long had access to rich data, but using it quickly—and responsibly—has been the challenge,” said Josh Walsh, CEO of BranchLab. “BranchLab solves this by turning privacy-safe, aggregated data into real-time insights that connect directly to activation. Teams can move faster, reach the right audiences more effectively, and drive better outcomes without relying on sensitive or individual-level information.”
At the core of the platform is a novel transformer-based architecture that enables models to train on health data within customer-controlled environments, while deploying only non-sensitive demographic and media-side signals. The new approach transforms commercialization workflows that used to take months and collapses them to minutes.
“The more we learned about BranchLab, the more we saw the possibility to transform the way patients learn about and get started on the right therapies,” said Carrie Williams, partner at McKesson Ventures. “Combining a deep understanding of the patient journey with a future-proofed approach to patient activation could enable an evolved patient therapy experience, one that seamlessly bridges privacy-first audience targeting with patient access, affordability and preference.”
FCA emphasized the underlying system shift: “Pharma has spent years investing in data and analytics, but much of it still lives outside of production systems,” said Andrew Bouldin, Managing Partner at FCA Venture Partners. “BranchLab’s approach to running models within regulated environments and connecting them directly to execution is a fundamental shift in how commercialization can work. We see this as a meaningful step toward making pharma’s data, and activation systems work together in real time.”
Sanofi Ventures highlighted the broader industry shift toward embedded, privacy-aware AI systems.
“Healthcare is entering a new era of responsible AI within regulated environments,” said Cris De Luca, Sanofi Ventures. “BranchLab has the potential to become a core commercial intelligence layer, putting this capability directly in the hands of pharma teams to connect therapies with patients across next‑generation digital environments.”
As the industry moves toward faster, more accountable, and privacy-first systems, BranchLab is positioning itself as the infrastructure layer for how therapies are brought to market in the AI era.
With the new funding, BranchLab will expand enterprise deployments, deepen integrations across the healthcare and media ecosystem, and continue building the unified AI platform for pharma commercialization.
About BranchLab
BranchLab is transforming healthcare commercialization with AI-native technology that helps leading healthcare organizations and their agencies connect patients, providers, and caregivers to life-improving therapies and resources. Built for the evolving privacy landscape, BranchLab improves commercialization performance without relying on individual-level health information. By modernizing the industry’s technical infrastructure, BranchLab helps organizations operate more efficiently and responsibly, while enabling a healthier system for everyone.
To learn more, visit www.branchlab.com.
CONTACT:
BranchLab Media Team
pr@branchlab.com

- Led by OrbiMed, with participation from RA Capital Management, Janus Henderson Investors, Sanofi Ventures, Harbour BioMed, and existing investors
- Proceeds will advance lead candidate WIN378 into Phase 3 and WIN027 into respiratory and dermatology studies by Q4 2026
- WIN378 has the potential to be the first-to-market, ultra long-acting anti-TSLP antibody for asthma and COPD • WIN027 is a long-acting bispecific targeting TSLP and IL-13 with best-in-disease efficacy potential
BASEL, Switzerland, May 4, 2026 (GLOBE NEWSWIRE) — Windward Bio, a private, clinical-stage biotechnology company committed to improving outcomes for people living with serious immunological diseases, today announced an upsized $165M crossover financing led by OrbiMed, with participation from existing Series A investors including Novo Holdings, Blue Owl Healthcare Opportunities, SR One, Omega Funds, RTW Investments, Qiming Venture Partners, Quan Capital, and Pivotal bioVenture Partners. The financing also included new investors RA Capital Management, Janus Henderson Investors, Sanofi Ventures, and Harbour BioMed. Proceeds will significantly extend the company’s cash runway and enable multiple clinical readouts in the next 12 months.
Since launching in January 2025, Windward Bio has in-licensed 2 clinical-stage assets, raised $365M, and rapidly advanced both programs in the clinic.
WIN378, the lead program, is a next-generation, fully human monoclonal antibody that potently binds to the thymic stromal lymphopoietin (TSLP) ligand. This well-validated cytokine plays a key role in the development and progression of a wide array of immunological diseases. WIN378 has the potential to be the first-to-market, ultra long-acting anti-TSLP antibody with twice-yearly dosing. The financing will accelerate the development of WIN378, which is currently being studied in the Phase 2/3 POLARIS program in asthma. The Phase 2 dose- ranging component of POLARIS is fully recruited, with initial data expected in second half of 2026. The first Phase 3 study of WIN378 is expected to begin in the fourth quarter of 2026. The Phase 2 SIRIUS study in chronic obstructive pulmonary disease (COPD) is anticipated to start in the second quarter of 2026.
WIN027, the second program, is a highly potent, long-acting bispecific antibody targeting both TSLP and interleukin-13 (IL-13), 2 well-validated and synergistic drivers of inflammation in severe asthma, COPD, and atopic dermatitis. WIN027 is currently in a Phase 1 study with data readout expected by the end of 2026. The financing will support multiple proof-of-concept studies across respiratory and dermatology indications starting in Q4 2026.
“We are excited to expand our shareholder base of top-tier investors to include RA Capital, Janus Henderson Investors, and Sanofi Ventures” said Luca Santarelli, MD, Founder, Chief Executive Officer, and Board Chair of Windward Bio. “This financing further strengthens our balance sheet and allows us to advance our programs of next-generation therapies for patients living with serious respiratory and dermatological diseases. ”
“Windward Bio is uniquely positioned to make an impact in immunology with a differentiated portfolio that could raise the standards of care in respiratory and dermatology,” said David Bonita, General Partner at OrbiMed.
“We are impressed with the significant progress the Windward Bio team has made since the company’s launch in 2025 and are proud to lead this high-caliber investor syndicate in supporting Windward Bio’s mission to discover and develop novel therapeutics for serious immunological conditions.”
About WIN378
WIN378 is a next-generation, fully human monoclonal antibody that potently inhibits the TSLP ligand. This clinically validated target plays a key role in the development and progression of a wide array of immunological diseases, including asthma and COPD. WIN378 has been engineered to achieve half-life extension (HLE) and have a silenced effector function. It has been studied in a Phase 1 trial, which confirmed an extended half-life suitable for twice-yearly dosing, demonstrated a low rate of antidrug antibodies, and was safe and well tolerated up to the highest dose tested. WIN378 is administered subcutaneously. Windward Bio licensed the global rights (excluding Greater China and several Southeast and West Asian countries) for WIN378 from Kelun-Biotech (also known as SKB378) and Harbour BioMed (also known as HBM9378). WIN378 is currently being evaluated in the POLARIS Phase 2/3 asthma study with initial readouts expected in the second half of 2026. A Phase 2 study in COPD is anticipated to begin in the second quarter of 2026. The first Phase 3 study of WIN378 is expected to begin in the fourth quarter of 2026.
About WIN027
WIN027 is a potential best-in-class, humanized bispecific monoclonal antibody with subpicomolar affinity for TSLP and IL-13, well-validated targets in immunological conditions. It has been engineered to achieve an extended half-life and enable less frequent dosing. Through this dual, long-acting inhibition, WIN027 is designed to set a new standard of efficacy in conditions such as asthma, COPD, and atopic dermatitis, potentially delivering deeper and more durable disease control than existing biologics. WIN027 is currently in Phase 1. Windward Bio licensed global rights (excluding Greater China) for WIN027 from Qyuns Therapeutics (also known as QX027N).
About Windward Bio
Windward Bio is a clinical-stage biotechnology company with deep discovery, development, and commercialization expertise committed to transforming the treatment of people living with serious immunological conditions. Its lead program is WIN378, a potential best-in-disease, ultra long-acting anti-TSLP monoclonal antibody currently in a Phase 2/3 trial for asthma. The pipeline also includes WIN027, a clinical- stage, long-acting anti-TSLPxIL-13 bispecific with broad therapeutic potential across immunological diseases, which is currently in Phase 1. The company is building a discovery pipeline of long-acting bispecific antibodies, targeting validated biology in respiratory and dermatological conditions.
Contacts
For media, business, or investor inquiries, please contact: media@windwardbio.com
VGA039 is a once monthly, subcutaneously self-administered investigational therapy for the treatment of bleeding disorders, initially being evaluated for von Willebrand disease (VWD)
Pivotal Phase 3 trial of VGA039 in adolescent and adult patients with VWD currently enrolling
SOUTH SAN FRANCISCO, CA, April 21, 2026 – Star Therapeutics, a late clinical-stage biotechnology company focused on the discovery and development of life-changing therapies for diseases with significant unmet need, today announced that the U.S. Food and Drug Administration (FDA) has granted rare pediatric disease designation (RPDD) and Breakthrough Therapy designation (BTD) to Star’s lead program, VGA039, for routine prophylaxis to prevent or reduce the frequency of bleeding episodes in patients with von Willebrand disease (VWD). VWD, the most common inherited bleeding disorder in which the blood does not clot properly, is a serious and life-threatening disease. VGA039 is a differentiated monoclonal antibody therapy that targets Protein S, with dual actions promoting platelet attachment and enhancing fibrin deposition to restore hemostasis.
“People living with VWD may face serious health complications, including frequent bleeding and hospitalizations that can significantly impact their quality of life. The FDA’s decision to grant both RPDD and BTD to VGA039 underscores the urgent need for new treatments for these patients, as well as Star’s potential to make a meaningful difference,” said Gary Patou, M.D., Chief Medical Officer of Star Therapeutics. “VGA039 could be transformative for patients, as it is designed to prevent or reduce bleeding across all types of VWD while reducing treatment burden via its once monthly, subcutaneous dosing regimen. We are excited to continue to partner with physicians, patients and advocacy organizations to enroll people aged 12 and over with all types of VWD in our ongoing, pivotal Phase 3 study evaluating VGA039 prophylaxis. We aim to bring this new treatment to people with VWD as efficiently as possible.”
The FDA’s RPDD is intended to encourage the development of therapies for serious or life- threatening rare diseases that primarily affect individuals aged 18 years or younger and impact fewer than 200,000 people in the U.S. Upon approval of a Biologics License Application (BLA), this designation makes Star eligible to receive a Priority Review Voucher (PRV), which may be redeemed to obtain priority review for a subsequent marketing application or transferred or sold to another sponsor. According to the National Organization for Rare Disorders, as of 2025, approximately 63 rare pediatric disease PRVs had been awarded across 47 conditions since the program began in 2012.
Building upon the Fast Track designation that VGA039 was awarded in 2025, the FDA’s BTD aims to expedite the development and review of therapies intended to treat serious conditions and address unmet medical needs. This designation is supported by interim data from the Phase 1/2 multidose study of VGA039 in adult and adolescent patients with VWD. These data were presented at the 67th American Society of Hematology (ASH) Annual Meeting and Exposition in December 2025, demonstrating substantial bleed reductions across all types of VWD and all types of bleeds.
About VGA039
VGA039 is an investigational monoclonal antibody therapy with a novel mechanism of action that targets Protein S, with dual actions promoting platelet attachment and enhancing fibrin deposition to restore hemostasis. VGA039 has the potential to be a universal hemostatic therapy that can treat numerous bleeding disorders, starting with all types of von Willebrand disease (VWD). As a subcutaneously self-administered investigational antibody therapy with a convenient once monthly dosing regimen, VGA039 has the potential to meaningfully improve convenience and quality of life for patients. VGA039 has received Fast Track, orphan drug, rare pediatric disease and Breakthrough Therapy designations from the U.S. Food and Drug Administration (FDA). VGA039 has advanced into a Phase 3 study (NCT07115004), VIVID-6, a global single arm cross-over study designed to investigate the safety and efficacy of subcutaneous administration of VGA039 as prophylaxis for bleeding in patients with every type of VWD. For additional information on our VIVID trials of VGA039, including how to enroll, please visit the website here.
About von Willebrand disease
Von Willebrand disease (VWD) is the most common inherited bleeding disorder in which the blood does not clot properly, caused by low or defective von Willebrand factor (VWF). VWD patients may experience excessive bleeding with varying severity and frequency, negatively impacting their daily lives. Current therapies for VWD prophylaxis include factor replacement therapies requiring multiple intravenous (IV) infusions every week. VWD affects more than 134,000 patients in the United States.
About Star Therapeutics
Star Therapeutics is a late-stage biotechnology company focused on the discovery and development of life-changing therapies for diseases with significant unmet need. Star’s leadership team brings deep expertise in antibody discovery, development, commercialization and strategic M&A, with a proven record of value creation across multiple biotechnology companies. For more information, please visit Star-Therapeutics.com and follow us on LinkedIn and X.
Contacts:
Media:
1AB
Katie Engleman
katie@1abmedia.com
Investors:
Star Therapeutics
Scott Robertson
scott@star-therapeutics.com

MinervaX Strengthens Executive Leadership Team with Two Strategic Appointments as it Advances its Maternal GBS Vaccine Towards Pivotal Trial Initiation
- The appointments of Jamila Louahed as CDO and Hans Henrik Chrois Christensen as CFO bring late‑stage development, global regulatory and operational expertise as MinervaX enters its next phase of growth
- Builds on robust Phase II results, paving the way for a registrational trial and advancing efforts to meet the urgent global need for a GBS vaccine
Copenhagen, Denmark, April 15, 2026 – MinervaX ApS, a privately held Danish biotechnology company developing a novel, prophylactic vaccine against Group B Streptococcus (GBS), today announces the appointments of Jamila Louahed as Chief Development Officer and Hans Henrik Chrois Christensen as Chief Financial Officer. These appointments strengthen MinervaX’s capabilities as the company advances its novel GBS vaccine toward pivotal trial initiation in the maternal indication and continues to scale its operations.
Jamila Louahed joins from GSK, where she held a senior leadership role in Vaccine Research and Development. She brings over 20 years of experience advancing vaccines from discovery through late-stage development and regulatory approval, with a strong track record in global program execution.
Hans Henrik Chrois Christensen brings more than two decades of financial leadership in the life science sector. He has served as CFO in both private and public companies, successfully leading financing strategies and M&A transactions, and supporting organizational growth and operational scale-up.
Per Fischer, Chief Executive Officer of MinervaX, said: "Jamila and Hans Henrik join us at an important moment for the company as we prepare for the initiation of our pivotal trial. Their combined expertise in vaccine development and financial strategy will strengthen our ability to execute and deliver on our long- term ambitions. We are united in our commitment to advancing a first‑in‑class GBS vaccine that has the potential to prevent life‑threatening infections affecting families worldwide."
Jamila Louahed, incoming Chief Development Officer of MinervaX, commented: "MinervaX is advancing one of the most promising approaches to GBS. The science is robust, the data are strong, and the program is ready for its next stage of development. Contributing to a vaccine with the potential to reshape outcomes for mothers, infants and older adults is a compelling opportunity."
Hans Henrik Chrois Christensen, incoming Chief Financial Officer of MinervaX, remarked: "MinervaX is at a pivotal point, having made solid progress towards starting its Pivotal trial in the maternal indication, supported by strong advisors and internationally renowned investors. Building the financial and operational strength required at this stage is critical to delivering the first approved GBS vaccine to patients worldwide."
These appointments build on a period of strong momentum for MinervaX. The Company has generated encouraging Phase II clinical data from its maternal immunization program, supported by preliminary surrogate markers of efficacy from natural history studies. Its protein‑only vaccine, based on fusions of highly immunogenic domains from the Alpha‑like protein family (AlpN), is designed to provide extremely broad protection against virtually all circulating GBS strains. Alongside its maternal program, MinervaX is also advancing development of its vaccine in older adults, having recently completed Phase I. These achievements, together with increasing global awareness of the significant burden of GBS, underscore the urgency of advancing a safe and effective vaccine into late‑stage trials.
Currently, there are no approved vaccines available for GBS, and the disease remains a major global cause of maternal and infant illness, amounting to an estimated 410,000 cases, and 147,000 stillbirths and infant deaths each year. This rising burden highlights the global importance of bringing an effective vaccine to patients as rapidly as possible.
For further information, please contact:
MinervaX
Per Fischer | Chief Executive Officer
Email: info@minervax.com
Optimum Strategic Communications Zoe Bolt | Vareen Outhonesack | Henry Williams
Email: minervax@optimumcomms.com
Tel: +44 (0) 203 882 9621
Notes to Editors:
MinervaX is a Danish biotechnology company, established in 2010 to develop a prophylactic vaccine against Group B Streptococcus (GBS), based on research from Lund University. MinervaX is developing a GBS vaccine for maternal immunization, and also for vaccination of older/at risk adults. Phase II clinical data from its maternal vaccination program suggest a high efficacy, based on the preliminary surrogate efficacy marker data from a natural history study. MinervaX’s GBS vaccine is a protein-only vaccine based on fusions of highly immunogenic and protective protein domains from selected surface proteins of GBS, the Alpha-like protein family (AIpN). Given the broad distribution of proteins contained in the vaccine on GBS strains globally, it is expected that MinervaX’s GBS vaccine will confer protection against virtually 100% of all GBS isolates.
Details of MinervaX’s completed Phase II clinical trials in pregnant people can be found at https://clinicaltrials.gov/ under the identifiers NCT04596878 and NCT05154578. In addition to pregnant people, MinervaX has also completed a Phase I trial of its novel GBS vaccine in older adults, under identifier NCT05782179. www.minervax.com
Combined company to operate as Korsana Biosciences and to advance Korsana’s potential best-in-class therapeutics to treat neurodegenerative diseases
Korsana’s lead program KRSA-028 is a next-generation shuttled monoclonal antibody targeting amyloid beta for the treatment of Alzheimer’s disease
Concurrent private financing of approximately $380 million expected to fund Korsana’s operations into 2029, including multiple clinical trial data readouts expected in 2027
Conference call scheduled for April 1, 2026, at 8:00 am ET
WALTHAM, Mass.--(BUSINESS WIRE)--Apr. 1, 2026-- Cyclerion Therapeutics, Inc. (“Cyclerion”) (Nasdaq: CYCN) and Korsana Biosciences, Inc. (“Korsana”), a privately-held biotechnology company discovering and developing novel therapies to reduce the burden of neurodegenerative diseases, today announced that they have entered into a definitive merger agreement for an all-stock transaction. Upon completion of the transaction, the combined company plans to operate under the name Korsana Biosciences, Inc. and trade on Nasdaq under the ticker symbol “KRSA.”
This press release features multimedia. View the full release here: https://www.businesswire.com/news/home/20260401287952/en/
In support of the proposed merger, Korsana has secured commitments for an oversubscribed private investment that is expected to result in total gross proceeds of approximately $380 million from a syndicate of investors led by Fairmount and Venrock Healthcare Capital Partners, with participation from General Atlantic, TCGX, Forbion, Wellington Management, Commodore Capital, RA Capital Management, RTW Investments, Vivo Capital, Janus Henderson Investors, Foresite Capital, J.P. Morgan Life Sciences Private Capital, SR One, Sanofi Ventures, Kalehua Capital, Spruce Street Capital, and other leading investment management firms. The financing includes common stock and pre-funded warrants exercisable for shares of Korsana common stock.
The financing is expected to close immediately prior to completion of the proposed merger. The combined company’s cash and cash equivalents balance at closing, including the funds from the private placement, is anticipated to fund Korsana’s operations into 2029 and provides runway through key clinical milestones. These include the advancement of KRSA-028 through Phase 1 healthy volunteer data expected in mid-2027 and interim proof of concept data measuring amyloid plaque clearance in Alzheimer’s patients expected by the end of 2027.
“Korsana is determined to deliver breakthrough medicines for patients suffering from neurodegenerative disorders. With our seasoned team and support from leading biotechnology investors, Korsana is well-positioned to advance a pipeline of innovative, next generation therapies,” said Jonathan Violin, Ph.D., Korsana’s President and Chief Executive Officer. “Patients deserve better options than what is currently available, and we believe our lead program KRSA-028 can deliver a best-in-class product to treat Alzheimer’s disease. We are also building a broader pipeline leveraging our proprietary platform to target other devastating neurodegenerative disorders.”
Korsana is the seventh company to launch with assets discovered and developed by Paragon Therapeutics. Korsana’s lead program is KRSA-028, a next-generation shuttled antibody targeting amyloid beta for the treatment of Alzheimer’s disease, discovered in partnership with Paragon Therapeutics. KRSA-028 utilizes the proprietary Therapeutic Targeting (THETA™) platform, which incorporates clinically validated transferrin receptor (TfR1) and Fc engineering and is designed to improve brain delivery and solve limitations of other TfR1-based approaches. KRSA-028 was designed to increase amyloid plaque clearance, reduce the rate of amyloid-related imaging abnormalities (ARIA) and hematologic adverse events, and optimize convenience and compliance with a low-volume subcutaneous route of administration. Korsana is advancing a pipeline of innovative THETA-enabled therapies for other undisclosed, neurodegenerative diseases with high unmet need.
“Our transaction with Korsana is the result of a comprehensive strategic review, and we believe it represents the best path forward for Cyclerion,” said Regina Graul, Ph.D., President and Chief Executive Officer of Cyclerion. “Korsana’s promising and innovative pipeline targeting neurodegenerative disorders, beginning with Alzheimer’s disease, provides the potential for significant value creation for Cyclerion’s shareholders.”
About the Proposed Transactions
Under the terms of the merger agreement, as of the closing of the proposed merger, the pre-merger Cyclerion shareholders are expected to own approximately 1.5% of the combined company and the pre-merger Korsana stockholders (inclusive of those investors participating in the pre-closing financing) are expected to own approximately 98.5% of the combined company. The percentage of the combined company that Cyclerion’s shareholders will own as of the closing of the proposed merger is subject to adjustment based on the estimated amount of Cyclerion’s net cash immediately prior to the closing date.
The transaction has received approval by the Board of Directors of both companies and is expected to close in the third quarter of 2026, subject to certain closing conditions, including, among others, approval by the stockholders of each company, the effectiveness of a registration statement to be filed with the U.S. Securities and Exchange Commission (the “SEC”) to register the securities to be issued in connection with the proposed merger, expiration of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, as amended, and the satisfaction of other customary closing conditions.
The combined company plans to operate under the name Korsana Biosciences, Inc. and will be led by Dr. Violin, Korsana’s current Chief Executive Officer. Korsana’s existing Board of Directors will become directors of the combined company, chaired by Tomas Kiselak, Founding Partner at Fairmount, and including Andrew Gottesdiener, M.D., Partner at Venrock Healthcare Capital Partners, Nilesh Kumar, Ph.D., Head of Biotech Private Investments at Wellington Management, Michelle Pernice, Operating Partner at Fairmount, Nimish Shah, Partner at Venrock Healthcare Capital Partners, and Dr. Violin.
Wedbush Securities Inc. is serving as exclusive strategic financial advisor and Gibson, Dunn & Crutcher LLP is serving as legal counsel to Korsana. Jefferies, TD Cowen, Stifel, UBS Investment Bank, and Wedbush & Co., LLC are serving as the placement agents to Korsana. Cooley LLP is serving as legal counsel to the placement agents. Gemini Valuation Services, LLC is serving as financial advisor and Ropes & Gray LLP is serving as legal counsel to Cyclerion.
Conference Call Details
The companies will hold a joint conference call on April 1, 2026, at 8:00 am ET to discuss the details of the proposed transactions.
Please access the presentation by clicking on the following link: https://register-conf.media-server.com/register /BIdbeaaec6359f483ab6e6685bd93e6312
About KRSA-028
KRSA-028 is an investigational, shuttled antibody targeting amyloid beta designed to advance the future of Alzheimer’s treatment. KRSA-028 utilizes a proven mechanism of action, selectively binding forms of amyloid beta that are enriched in amyloid plaques. KRSA-028 leverages Korsana’s Therapeutic Targeting (THETA™) technology, which incorporates clinically validated transferrin receptor (TfR1) and Fc engineering and is designed to improve brain delivery, safety, and convenience.
About Cyclerion Therapeutics
Cyclerion is a biopharmaceutical company focused on developing treatments for neuropsychiatric diseases. Cyclerion’s foundational product candidate, CYC-126, is an individualized therapy for treatment-resistant depression, a condition with significant unmet medical need and substantial commercial opportunity.
For more information about Cyclerion, please visit https://www.cyclerion.com/ and follow us on X (@Cyclerion) and LinkedIn (www.linkedin.com/company/cyclerion).
About Korsana Biosciences
Korsana Biosciences was founded to reduce the burden of neurodegenerative diseases for patients and caregivers. Developed in partnership with Paragon Therapeutics, Therapeutic Targeting (THETA™) is a novel blood-brain barrier-penetrant shuttle platform designed to enable dramatically higher drug concentration inside the brain and overcome the limitations of earlier shuttle technologies. Korsana is advancing a pipeline of innovative THETA-enabled therapies for neurodegenerative diseases, starting with KRSA-028 for the treatment of Alzheimer’s disease. For more information, please visit www.korsana.com.
Forward-Looking Statements
This press release contains forward-looking statements concerning Cyclerion, Korsana, the proposed pre-closing financing and the proposed merger (collectively, the “Proposed Transactions”) and other matters. These forward-looking statements include, but are not limited to, express or implied statements relating to Cyclerion’s and Korsana’s management teams’ expectations, hopes, beliefs, intentions or strategies regarding the future including, without limitation, statements regarding: the Proposed Transactions and their expected effects, perceived benefits or opportunities, including expected investment amounts and proceeds from investors, and related timing with respect thereto; expectations regarding or plans for discovery, preclinical studies, clinical trials and research and development programs, in particular with respect to KRSA-028, and any developments or results in connection therewith; the anticipated timing of the commencement of and results from those studies and trials; expectations regarding the use of proceeds, the sufficiency of post-transaction resources to support the advancement of Korsana’s pipeline through certain milestones and the time period over which the combined company’s post-transaction capital resources will be sufficient to fund the combined company’s anticipated operations; the combined company operating under the name Korsana Biosciences, Inc. and trading on Nasdaq under the ticker symbol “KRSA”; the expected ownership percentages of pre-merger Cyclerion and Korsana stockholders following the closing of the proposed merger; the potential for KRSA-028 to be a best-in-class treatment for Alzheimer’s disease; and the anticipated advancement of Korsana’s broader pipeline of THETA-enabled therapies for neurodegenerative diseases. The words “opportunity,” “potential,” “milestones,” “pipeline,” “can,” “goal,” “strategy,” “target,” “anticipate,” “achieve,” “believe,” “contemplate,” “continue,” “could,” “estimate,” “expect,” “intends,” “may,” “plan,” “possible,” “project,” “should,” “will,” “would” and similar expressions (including the negatives of these terms or variations of them) may identify forward-looking statements, but the absence of these words does not mean that a statement is not forward-looking. All statements contained in this press release that do not relate to matters of historical fact should be considered forward-looking statements.
These forward-looking statements are based on management’s current expectations and assumptions as of the date of this press release and are subject to a number of known and unknown risks, uncertainties, and other factors that could cause actual results to differ materially from those expressed or implied by such statements, including, without limitation, the following: the risk that the Proposed Transactions may not be completed on the anticipated timeline or at all; the failure to satisfy the conditions to the closing of the merger, including obtaining the requisite approvals of the stockholders of each of Cyclerion and Korsana and the effectiveness of the registration statement to be filed with the SEC in connection with the Proposed Transactions; risks related to the clinical development of KRSA-028, including the possibility of delays, unfavorable clinical results, safety or tolerability issues, or the failure to obtain regulatory approval; uncertainties regarding the capabilities and potential of the THETA platform and Korsana’s pipeline programs; the risk that the financing may not close or may not generate the anticipated proceeds; market, macroeconomic, or other conditions that could adversely affect the combined company’s cash runway or ability to raise additional capital; risks related to the integration of the two companies and the management of a newly public company; and the highly competitive nature of the Alzheimer’s disease and neurodegenerative disease therapeutic landscape, including the risk that competitors may develop superior or more cost-effective therapies.
Additional factors that may cause actual results to differ materially from those expressed or implied by the forward-looking statements in this press release are discussed in Cyclerion’s filings with the SEC, including its most recent Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, and other reports filed with the SEC from time to time, and will be discussed in the registration statement to be filed by Cyclerion with the SEC in connection with the Proposed Transactions. Readers are cautioned not to place undue reliance on these forward-looking statements. Each of Cyclerion and Korsana expressly disclaims any obligation to update or revise any forward-looking statements, whether as a result of new information, future events, or otherwise, except as required by applicable law. All forward-looking statements are made as of the date of this press release.
No Offer or Solicitation
This press release and the information contained herein is not intended to and does not constitute (i) a solicitation of a proxy, consent or approval with respect to any securities or in respect of the Proposed Transactions or (ii) an offer to sell or the solicitation of an offer to subscribe for or buy or an invitation to purchase or subscribe for any securities pursuant to the Proposed Transactions or otherwise, nor shall there be any sale, issuance or transfer of securities in any jurisdiction in contravention of applicable law. No offer of securities shall be made except in accordance with the requirements of the Securities Act of 1933, as amended, or an exemption therefrom. Subject to certain exceptions to be approved by the relevant regulators or certain facts to be ascertained, no public offer will be made directly or indirectly, in or into any jurisdiction where to do so would constitute a violation of the laws of such jurisdiction, or by use of the mails or by any means or instrumentality (including without limitation, facsimile transmission, telephone and the internet) of interstate or foreign commerce, or any facility of a national securities exchange, of any such jurisdiction.
NEITHER THE SEC NOR ANY STATE SECURITIES COMMISSION HAS APPROVED OR DISAPPROVED OF THE SECURITIES OR DETERMINED IF THIS PRESS RELEASE IS TRUTHFUL OR COMPLETE.
Important Additional Information about the Proposed Transactions Will be Filed with the SEC
This press release is not a substitute for the registration statement or for any other document that Cyclerion may file with the SEC in connection with the Proposed Transactions. In connection with the Proposed Transactions between Cyclerion and Korsana, Cyclerion intends to file relevant materials with the SEC, including a registration statement on Form S-4 that will contain a proxy statement/prospectus of Cyclerion. CYCLERION URGES INVESTORS AND SHAREHOLDERS TO READ THE REGISTRATION STATEMENT, PROXY STATEMENT/PROSPECTUS AND ANY OTHER RELEVANT DOCUMENTS THAT MAY BE FILED WITH THE SEC, AS WELL AS ANY AMENDMENTS OR SUPPLEMENTS TO THESE DOCUMENTS, CAREFULLY AND IN THEIR ENTIRETY IF AND WHEN THEY BECOME AVAILABLE BECAUSE THEY WILL CONTAIN IMPORTANT INFORMATION ABOUT CYCLERION, KORSANA, THE PROPOSED TRANSACTIONS AND RELATED MATTERS. Investors and shareholders will be able to obtain free copies of the proxy statement/prospectus and other documents filed by Cyclerion with the SEC (when they become available) through the website maintained by the SEC at www.sec.gov. Shareholders are urged to read the proxy statement/prospectus and the other relevant materials when they become available before making any voting or investment decision with respect to the Proposed Transactions. In addition, investors and shareholders should note that Cyclerion communicates with investors and the public using its website (www.cyclerion.com).
Participants in the Solicitation
Cyclerion, Korsana and their respective directors and executive officers may be deemed to be participants in the solicitation of proxies from shareholders in connection with the Proposed Transactions. Information about Cyclerion’s directors and executive officers, including a description of their interests in Cyclerion, is included in Cyclerion’s most recent Annual Report on Form 10-K, subsequent Quarterly Reports on Form 10-Q filed with the SEC, including any information incorporated therein by reference, as filed with the SEC, and other documents that may be filed from time to time with the SEC. Additional information regarding these persons and their interests in the transaction will be included in the proxy statement/prospectus relating to the Proposed Transactions when it is filed with the SEC. These documents can be obtained free of charge from the sources indicated above.
View source version on businesswire.com: https://www.businesswire.com/news/home/20260401287952/en/
For Korsana Investor and Media Inquiries:
info@korsana.com
For Cyclerion Investor and Media Inquiries:
IR@cyclerion.com
Source: Korsana Biosciences, Inc. and Cyclerion Therapeutics, Inc.
Boston, MA, March 25, 2026 – NodThera, the leading clinical-stage NLRP3 company developing a portfolio of best-in-class brain-penetrant and immune-targeted oral inhibitors to address cardiometabolic and neuroinflammatory diseases driven by the NLRP3/IL-6/IL-1 inflammation pathway, today announced participation in the following investor conferences:
- Piper Sandler 3rd Annual Virtual Cardio Day
April 1, 2026
Virtual - 25th Annual Needham Virtual Healthcare Conference
April 13, 2026
Virtual - Piper Sandler Spring Biopharma Symposium
April 15, 2026
Boston, MA
About NLRP3
NLRP3 is an upstream activator of proinflammatory cytokines, including IL-6, IL-18 and IL-1β. Chronic activation of NLRP3 drives pathologic inflammation in cardiometabolic, neurological and other diseases. NLRP3 inhibition with targeted oral therapies reduces systemic inflammation to a similar degree as biologics and does not cause immunosuppression.
About NodThera
NodThera is the leading clinical-stage NLRP3 company developing a portfolio of best-in-class brain-penetrant and immune-targeted oral inhibitors to address unmet needs in cardiometabolic and neuroinflammatory diseases driven by the NLRP3/IL-6/IL-1 inflammation pathway.
NodThera is backed by top-tier investors including Blue Owl Capital, Novo Holdings, F-Prime Capital, 5AM Ventures, Sofinnova Partners, Epidarex Capital, and Sanofi Ventures.
NodThera is headquartered in Boston, Massachusetts, with an R&D base in the UK.
Learn more at www.nodthera.com or follow the Company on LinkedIn.
Investors and Media
Argot Partners
nodthera@argotpartners.com
-- The trial uses a response-driven dosing strategy and extended treatment across three prostate cancer populations, supported by the favorable clinical, pharmacokinetic and dosimetry results
from Phase 1 b --
Brisbane, Australia and Boston, USA — February 24, 2026 - AdvanCell, a clinical-stage radiopharmaceutical company developing innovative targeted alpha therapies for cancer, today announced the novel Phase 2 design of the ongoing TheraPb clinical trial of ADVC001 in metastatic prostate cancer. ADVC001 is an investigational Lead-212-based prostate-specific membrane antigen {PSMA)-targeted alpha therapy. The study design will be showcased in a 'Trials in Progress' poster this week at the American Society of Clinical Oncology Genitourinary Cancers Symposium (ASCO GU 2026) in San Francisco, CA.
The TheraPb Phase 2 is an open-label, randomized expansion study incorporating dose optimization strategies in three groups of patients with metastatic hormone-sensitive {mHSPC) and castration-resistant prostate cancer {mCRPC). The study's innovative design is supported by the Phase 1 b dose escalation results, which showed encouraging safety and promising anti- tumor activity of ADVC001.Atthe recommended Phase 2 dose, 80% of patients achieved a 50% reduction in prostate-specific antigen {PSA50), and a 100% overall response rate was observed in patients with evaluable tumors, while no dose-limiting toxicities and no toxicity-related treatment discontinuations or dose modifications were observed. ADVC001 exhibited rapid and high tumor uptake, fast renal clearance, and low normal-organ radiation exposure.
"The Phase 2 TheraPb trial employs a novel dosing strategy that incorporates more frequent up- fronttreatment leveraging the half-life of Lead-212, as well as taking into account patient-specific responses;• commented Thomas Hope, MD, Vice Chair Clinical Operations and Strategy, Department of Radiology and Biomedical Imaging, University of California San Francisco. "This approach is the first in the next generation of radioligand therapy trials that goes beyond standard dosing strategies in order to improve patient outcomes."
"ADVC001 is a promising alpha PSMA-directed theranostic treatment that uses an innovative payload of Lead-212" commented Michael J. Morris, MD, Prostate Cancer Section Head, Genitourinary Oncology, Memorial Sloan Kettering Cancer Center. "The study examines the merits of a treatment intensification strategy, adaptive dosing, and application across multiple clinical settings under the auspices of a novel study design intended to facilitate efficient development"
"The TheraPb Phase 2 incorporates dose optimization strategies designed to improve patient outcomes in three distinct prostate cancer populations across the disease continuum, with high unmet medical need:' said Anna Karmann, MD PhD, Chief Medical Officer at AdvanCell. "To maximize clinical benefit and minimize toxicity, we are investigating novel dosing regimens aiming to optimize timing and duration of treatment. Encouraged by the favorable safety and tolerability results of ADVC001 in Phase 1 b, the study also includes evaluation of extended treatment in participants who demonstrate ongoing benefit"
The TheraPb Phase 2 expansion is designed to optimize ADVC001 development opportunities. Participants with mHSPC (suboptimal responders), mCRPC (pre-chemotherapy) and mCRPC (post-177Lu-PSMA) are randomized to either 160 or 200 MBq of ADVC001 and receive up to 4 doses of ADVC001 during a dose-intense induction. Additional doses may be administered as maintenance treatment for a total of up to 12 doses, allowing for a treatment pause ('treatment holiday') based on individual response.
The TheraPb Phase 2 trial (NCT05720130) is open at clinical sites in Australia, with planned expansion to additional sites in the United States.
Trials in Progress Poster at ASCO GU 2026
• Presentation Title: Phase 2 Expansion Study of 212Pb-ADVC001 in Metastatic Prostate
Cancer: The TheraPb Trial
• Presenter: Professor Aaron Hansen, Princess Alexandra Hospital, Brisbane, Australia
• AbstractNumber:TPS280, Board N16
• Session: Trials in Progress Poster Session A: Prostate Cancer
• Date and Time: 26-February - 11 :30 AM - 12:45 PM and 5:45 PM - 6:45 PM Pacific Ti me
The presentation will be available on AdvanCell's website at www.advancell.com.au/presentations-publications
XXX
About 212Pb-ADVC001
212Pb-ADVC001 (ADVC001) is a proprietary and patented PSMA-targeting radioligand with optimized physicochemical properties and labelled with Lead-212 (212Pb), an alpha-emitting payload (radionuclide) with a high dose rate, 10.6-hour half-life and simple decay scheme. ADVC001 is designed to deliver radiation at a cellular level to effectively kill prostate cancer cells while minimizing toxicity.
About the Th era Pb trial
The TheraPb trial (NCT05720130) is a prospective, open-label Phase 1/2 dose escalation and expansion study evaluatingADVC001 in metastatic prostate cancer. The completed Phase 1 b dose escalation assessed the safety and tolerability of escalating doses of ADVC001 administered every 6, 4, 2 or 1 week(s) (see press release). The Phase 2 expansion is assessing the efficacy and safety of ADVC001 at two dose levels. The trial utilizes a randomized dose- response design and dose optimization elementstoevaluateADVC001 in PSMA-positivemCRPC and in mHSPC.
About AdvanCell
AdvanCell is a vertically integrated, clinical-stage radiopharmaceutical company dedicated to developing innovative cancer therapies that harness the power of targeted alpha-emitting radionuclides. By leveraging its proprietary Lead-212 platform, advanced and scalable manufacturing and world-class clinical development capabilities, AdvanCell aims to deliver novel treatments that improve outcomes for patients with cancer globally. For more information, visit www.advancell.com.au and follow us on Linkedln.
MSK disclosure: Dr. Morris has financial interests related to AdvanCell.
Contacts
Anna Karmann, CMO
contact@advancell.com.au
For media inquiries, please contact:
MEDiSTRAVA (in the UK)
advancell@medistrava.com
+44 (0)20 3928 6700
Boston, MA, February 23, 2026 – NodThera, the leading clinical-stage NLRP3 company developing a portfolio of best-in-class brain-penetrant and immune-targeted oral inhibitors to address cardiometabolic and neuroinflammatory diseases driven by the NLRP3/IL-6/IL-1 inflammation pathway, today announced that Geoff McDonough, M.D., Chief Executive Officer of NodThera, will present at the Oppenheimer 36th Annual Healthcare Life Sciences Conference being held February 25-26, 2026, in a virtual format.
Details of the presentation are as follows:
Oppenheimer 36th Annual Healthcare Life Sciences Conference
Date: Thursday, February 26, 2026
Time: 11:20 AM ET
Presenter: Geoff McDonough, M.D.
Location: virtual
About NLRP3
NLRP3 is an upstream activator of proinflammatory cytokines, including IL-6, IL-18 and IL-1β. Chronic activation of NLRP3 drives pathologic inflammation in cardiometabolic, neurological and other diseases. NLRP3 inhibition with targeted oral therapies reduces systemic inflammation to a similar degree as biologics and does not cause immunosuppression.
About NodThera
NodThera is the leading clinical-stage NLRP3 company developing a portfolio of best-in-class brain-penetrant and immune-targeted oral inhibitors to address unmet needs in cardiometabolic and neuroinflammatory diseases driven by the NLRP3/IL-6/IL-1 inflammation pathway.
NodThera is backed by top-tier investors including Blue Owl Capital, Novo Holdings, F-Prime Capital, 5AM Ventures, Sofinnova Partners, Epidarex Capital, and Sanofi Ventures.
NodThera is headquartered in Boston, Massachusetts, with an R&D base in the UK.
Learn more at www.nodthera.com or follow the Company on LinkedIn.
Investors and Media
Argot Partners
nodthera@argotpartners.com
Lead program KRSA-028 is a next-generation shuttled monoclonal antibody targeting amyloid beta for the treatment of Alzheimer’s disease
KRSA-028 preclinical data support potential for a best-in-class therapeutic profile enabled by THETA™, Korsana’s proprietary TfR1-based therapeutic targeting plaJorm
Korsana is led by veteran CEO Jonathan Violin, Ph.D., and is the seventh company launched based on assets discovered by Paragon Therapeutics
Company is capitalized through key clinical milestones in 2027, with cash runway into 2028
WALTHAM, Mass.,February [18], 2026—Korsana Biosciences, Inc. (“Korsana” or the “Company”), a biotechnology company discovering and developing novel therapies to reduce the burden of neurodegenerative diseases, today announced its emergence from stealth, with backing from a leading syndicate of healthcare investors. The Company was founded in 2024 with a $25 million seed investment from Fairmount and Venrock Healthcare Capital Partners. In September 2025, the Company closed a $150 million private Series A financing co-led by Wellington Management and TCGX, with participation by J.P. Morgan Life Sciences Private Capital, Janus Henderson Investors, Sanofi Ventures, Foresite Capital, and others. This funding is being used to develop potential best-in-class therapeutics to treat neurodegenerative diseases, with an initial focus on Alzheimer’s disease.
Korsana’s lead program is KRSA-028, a next-generation shualed antibody targeting amyloid beta for the treatment of Alzheimer’s disease, discovered in partnership with Paragon Therapeutics. KRSA-028 utilizes the proprietary Therapeutic Targeting (THETA™) pladorm, which incorporates clinically validated transferrin receptor (TfR1) and Fc engineering and is designed to improve brain delivery and solve limitations of other TfR1-based approaches. KRSA-028 was designed to increase amyloid plaque clearance, reduce the rate of amyloid-related imaging abnormalities (ARIA) and hematologic adverse events, and optimize convenience and compliance with a low-volume subcutaneous route of administration. Korsana is advancing a pipeline of innovative THETA-enabled therapies for other undisclosed, neurodegenerative diseases with high unmet need.
Korsana has appointed Jonathan Violin, Ph.D. as President and Chief Executive Officer. Dr. Violin has been a venture partner at Fairmount since 2023 and previously served as founding CEO for Viridian Therapeutics from 2020 to 2023. Dr. Violin was also the founding CEO of Dianthus Therapeutics and of Quellis Biosciences, which merged into Astria Therapeutics.
“We are thrilled to announce the launch of Korsana with a mission to elevate expectations for patients suffering from devastating neurodegenerative diseases,” said Dr. Violin. “In particular, Alzheimer’s disease represents a massive and growing unmet need, with the U.S. patient population projected to double to approximately 13 million by 2050. Only two disease-modifying therapies have been approved to treat Alzheimer’s, and both carry safety warnings, offer only modest efficacy, and impose a high burden of care. Patients deserve beaer options than what is currently available, and we believe our lead program KRSA- 028 can deliver a best-in-class product to treat Alzheimer’s. I am thrilled with the robust support of our mission by Korsana’s investors, and excited to build another leading biotechnology company in partnership with the team at Paragon Therapeutics.”
“We believe Korsana is well positioned to overcome the limitations of not just first-generation Alzheimer’s therapies, but also of earlier shuale technologies,” added Andrew Goaesdiener, M.D., partner at Venrock and co-Founder and Member of the Board of Directors of Korsana. “Leveraging a well-established regulatory pathway, Korsana is advancing the next generation of therapies for Alzheimer’s disease and other neurodegenerative disorders, with a clear path to creating value for patients. We are thrilled to work with Korsana to build what I believe will become an important and valuable company. Together, we are commiaed to delivering beaer medicines to patients suffering from neurodegenerative diseases.”
Korsana’s initial financing is expected to fund activities into 2028 and provides runway through key clinical milestones, which include potentially differentiating pharmacokinetics, CNS penetration, and safety data from healthy volunteers expected in mid-year 2027, and initial proof of concept data demonstrating amyloid plaque clearance in Alzheimer’s patients expected by the end of 2027.
“We are excited to come together with the rest of this investor syndicate to support Korsana’s launch,” said Nilesh Kumar, Head of Biotech Private Investments, Wellington Management and Member of the Board of Directors of Korsana. “We look forward to seeing KRSA-028 and Korsana’s pipeline of innovative therapies advance through key clinical milestones in the coming years.”
About KRSA-028
KRSA-028 is an investigational, shualed antibody targeting amyloid beta designed to advance the future of Alzheimer’s treatment. KRSA-028 utilizes a proven mechanism of action, selectively binding forms of amyloid beta that are enriched in amyloid plaques. KRSA-028 leverages Korsana’s Therapeutic Targeting (THETA™) technology, which incorporates clinically validated transferrin receptor (TfR1) and Fc engineering and is designed to improve brain delivery, safety, and convenience.
About Korsana Biosciences
Korsana Biosciences was founded to reduce the burden of neurodegenerative diseases for patients and caregivers. In partnership with Paragon Therapeutics, Korsana has developed Therapeutic Targeting (THETA™), a novel blood-brain barrier-penetrant shuale pladorm designed to enable dramatically higher drug concentration inside the brain and overcome the limitations of earlier shuale technologies. Korsana is advancing a pipeline of innovative THETA-enabled therapies for neurodegenerative diseases, starting with KRSA-028 for the treatment of Alzheimer’s disease. For more information, please visit www.korsana.com.
About Paragon Therapeutics
Paragon Therapeutics, Inc., is a biotechnology company applying cupng-edge science and technology to shape the next generation of novel best-in-class complex biologics for major medical needs. The company is fueling a pipeline of transformative therapies that are positioned to be rapidly advanced into clinical development by partner companies. To date, six companies have been launched based on assets developed by Paragon. Founded by Fairmount in 2021, Paragon is based in Waltham, MA. For more information, please visit www.paragontherapeutics.com. Follow us on LinkedIn.
CONTACT
For Investor and Media:
Argot Partners
212-600-1902
korsana@argotpartners.com

PRINCETON, NJ and SHANGHAI, CHINA – QuantX Biosciences, Inc., a computationally driven drug discovery and development company using advanced modeling and high-performance computing to rapidly design and optimize oral small molecule therapeutics, announced today the close of an oversubscribed $85 million Series B financing co-led by LAV and Sanofi Ventures. Additional investors included Hongshan, and existing investors.
The proceeds from this financing will be used to support the clinical development of its best-in-class STAT6 oral small molecule inhibitor and IL-17 oral small molecule inhibitor, in addition to continued discovery efforts for oral therapies in immunology and inflammation.
“This fundraising is a watershed moment for QuantX as we continue to refine our strong discovery engine, add to our robust pipeline, and advance multiple programs towards human studies,” said Wei Li, Ph.D., Interim Chief Executive Officer of QuantX and a Founding Partner of Creacion Ventures. “In only three years, we’ve assembled a world-class team that’s been successful in not only discovering multiple, new, first-in-class small molecule candidates for diseases with large patient populations, but have also advanced these to IND-enabling studies.”
David Wang, MD, Ph.D., Partner and Senior Managing Director of OrbiMed, added, “The combination of QuantX’s ability to deliver best-in-class assets and the speed by which they’re able to advance these programs makes for a unique value proposition across a variety of therapeutic indications. We look forward to continuing our support of QuantX as they endeavor to transform the standards of care across a range of diseases.”
QuantX Biosciences was co-founded by veteran drug hunters Wayne Tang, Ph.D. and Yax Sun, Ph.D. in 2022. Since its inception, the company has focused on the development of a world class computational drug discovery platform by merging physics-based and statistical modeling, which has resulted in the development of multiple high quality oral small molecule development candidates.
Previously incubated by OrbiMed and Creacion Ventures, QuantX has raised $130 million to date.
About QuantX
QuantX Biosciences is a privately held pharmaceutical company advancing oral small-molecule medicines through a computationally driven drug discovery platform. The company integrates physics-based modeling, statistical methods, and computational chemistry to rapidly design and optimize drug candidates against clinically validated targets. QuantX is focused on building a pipeline of differentiated therapies for immune-mediated and inflammatory diseases, with the goal of improving development efficiency and success rates. Headquartered in Princeton, New Jersey, QuantX Biosciences is backed by leading life science investors and a team with deep expertise in drug discovery and development.
QuantX Contact:
Peter Greensmith
QuantX Biosciences
peter.greensmith@quantbio.com
Media Contact
Andrew W. Mielach
LifeSci Communications
amielach@lifescicomms.com

Initiates first-in-human clinical studies of an IgG-like CD3 bispecific designed with a wide therapeutic index to selectively redirect T cells to B7-H7–expressing tumors, including lung adenocarcinoma, renal cell carcinoma and pancreatic adenocarcinoma
Clinical trial design with efficient dose escalation algorithm and biomarker strategy allows rapid data readout and enables optimized dosing and response
Cambridge, MA – February 9, 2026 – NextPoint Therapeutics, a clinical-stage biotechnology company developing a new world of precision therapeutics through its leading scientific work on the novel B7-H7 axis, today announced the clearance of an Investigational New Drug (IND) application with the U.S. Food and Drug Administration (FDA) to initiate clinical development of NPX372, a first-in-class T cell engager (TCE) for the treatment of patients with solid tumors.
B7-H7 is highly specific to tumor epithelial cells and largely absent from normal tissue, setting it apart as a much cleaner and more selective target for a T cell engager. NPX372 is an IgG-like bispecific T cell engager designed to drive target expression–proximal cytotoxicity while minimizing non-specific T cell activation. In preclinical studies, NPX372 demonstrated complete tumor regression in solid tumor models, while displaying great tolerability in relevant preclinical safety assessment and no evidence of cytokine release syndrome.
“T-cell engagers represent a novel approach to delivering sustained anti-tumor immune responses to cancer patients,” said Leena Gandhi, MD, PhD, Chief Medical Officer of NextPoint Therapeutics. “This IND clearance enables us to accelerate the delivery of targeted immune therapy to broad patient populations in need, including patients with lung adenocarcinoma, renal cell carcinoma and pancreatic adenocarcinoma. Our clinical program deploys an efficient dose escalation algorithm and utilizes a novel biomarker assay to select patients with the highest chance of benefit from a B7-H7 targeting TCE.”
“The NPX372 IND clearance represents the strategic initiation of our clinical use of B7-H7 as a highly specific tumor-targeting antigen for potent direct tumor killing therapeutics,” said Ivan Cheung, Chief Executive Officer of NextPoint Therapeutics. “The ideal expression profile of B7-H7 and the meticulous construct design of NPX372, along with a clinical trial design that enables fast and impactful data readouts, position NPX372 as a frontrunner in the emerging T cell engager field for solid tumors.”
About NextPoint Therapeutics
NextPoint is launching a new world of precision therapeutics through its leading scientific work on the novel B7-H7 axis. Our team of proven drug developers is advancing a T-cell engager with wide therapeutic window, an antibody-drug conjugate with our proprietary linker technology, and a multi-functional checkpoint inhibitor. Our innovative approach integrates foundational science with a defined clinical biomarker to identify the right patient population for each B7-H7-directed modality, so that we can deliver first-in-class therapies to a broad range of cancer patients with B7-H7 upregulation including those who do not benefit from currently approved therapies such as PD-1/L1 inhibitors. To learn more, visit nextpointtx.com.
Contacts
Media Contact
Lauren Arnold
LA Communications
Lauren@lacommunications.net
Dr. McDonough brings global company-building, commercial strategy, and R&D leadership experience to the Company ahead of key clinical milestones
NodThera plans to report results from two Phase 2 cardiometabolic trials RESOLVE-1 and RESOLVE-2 in mid-2026 and third quarter 2026 respectively
Company preparing to begin cardiometabolic Phase 3 trial in 1H 2027
Boston, MA, February 4, 2026 – NodThera, the leading clinical-stage NLRP3 company developing a portfolio of best-in-class brain-penetrant and immune-targeted oral inhibitors to address unmet needs in cardiometabolic and neuroinflammatory diseases driven by the NLRP3/IL-6/IL-1 inflammation pathway, announced the appointment of Geoff McDonough, M.D., as Chief Executive Officer. Dr. McDonough is a seasoned physician-executive with extensive global biopharma industry experience who joins NodThera’s management team ahead of a series of significant anticipated clinical milestones.
“The NLRP3/IL-6/IL-1 pathway underlies several large cardiometabolic and neurologic diseases with significant unmet need,” said Dr. McDonough. “NodThera’s unique brain penetrant and immune-targeted lead molecule, NT-0796, will complete two cardiometabolic Phase 2 trials shortly, and we are preparing to launch a Phase 3 trial in just over a year. These milestones are key to our mission to reach patients and families suffering from diseases driven by chronic inflammation.”
“Geoff’s insights and expertise will be invaluable as we approach an important series of clinical milestones as NT-0796 moves toward Phase 3 development for cardiometabolic diseases,” said Don Nicholson, Ph.D., Chair of the Board. “Geoff brings an impressive combination of global commercial, strategic and R&D leadership experience that positions him well to lead NodThera into this next phase of growth.”
Dr. McDonough brings more than two decades of leadership experience. Most recently, he served as President & Chief Executive Officer of Generation Bio (NASDAQ: GBIO). Dr. McDonough previously served as President & CEO of Sobi, overseeing substantial portfolio expansion and international growth. Earlier in his career, Dr. McDonough spent a decade at Genzyme, holding senior roles including President of Genzyme Europe and Senior Vice President & General Manager of the global Lysosomal Storage Diseases business. He trained in internal medicine and pediatrics at Massachusetts General Hospital and Boston Children’s Hospital and earned his M.D. from Harvard Medical School.
About RESOLVE-1 and RESOLVE-2 Phase 2 Clinical Trials
RESOLVE-1 is a cardiometabolic Phase 2 clinical trial designed to evaluate the safety and efficacy of NT-0796 over six months. This trial, the only NLRP3 trial to include patients with type 2 diabetes, evaluates improvements in systemic inflammation (high sensitivity CRP) and cardiometabolic markers, and is fully enrolled with 176 patients randomized to NT-0796 or placebo. This is the largest and longest clinical trial in this class and is on track to read out in mid-2026.
RESOLVE-2 is a cardiometabolic Phase 2 clinical trial designed to evaluate the safety and efficacy of NT-0796 in combination with a GLP- agonist. This fully-enrolled study will evaluate markers of systemic inflammation and cardiometabolic health and is on track to read out in 3Q 2026.
About NLRP3
NLRP3 is an upstream activator of proinflammatory cytokines, including IL-6, IL-18 and IL-1β. Chronic activation of NLRP3 drives pathologic inflammation in cardiometabolic, neurological and other diseases. NLRP3 Inhibition with targeted oral therapies reduces systemic inflammation to a similar degree as biologics and does not cause immunosuppression.
About NodThera
NodThera is the leading clinical-stage NLRP3 company developing a portfolio of best-in-class brain-penetrant and immune-targeted oral inhibitors to address unmet needs in cardiometabolic and neuroinflammatory diseases driven by the NLRP3/IL-6/IL-1 inflammation pathway.
NodThera is backed by top-tier investors including Blue Owl Capital, Novo Holdings, F-Prime Capital, 5AM Ventures, Epidarex Capital, Sofinnova Partners and Sanofi Ventures.
NodThera is headquartered in Boston, Massachusetts, with an R&D base in the UK.
Learn more at www.nodthera.com or follow the Company on LinkedIn.
Investors and Media
Argot Partners
nodthera@argotpartners.com
OXFORD, United Kingdom, Jan. 28, 2026 (GLOBE NEWSWIRE) -- OMass Therapeutics (‘OMass’ or ‘the Company’), a biotechnology company identifying medicines against highly validated target ecosystems such as membrane proteins or intracellular complexes, today announced updates to its Board of Directors, including the appointment of Jonathan Montagu, Chief Executive Officer and co-founder of HotSpot Therapeutics, as Board Chair. In conjunction with the appointment, existing Directors Jim Geraghty and Birgitte Volck will retire from the board and continue to support the company as strategic advisers.
“Over the past year, OMass has made meaningful progress across its portfolio, including advancing our lead MC2 antagonist towards the clinic, establishing in vivo efficacy for our first-in-class SLC15A4 program, and continuing to demonstrate the power of our OdyssION™ platform,” said Ros Deegan, Chief Executive Officer of OMass Therapeutics. “I would like to thank Jim and Birgitte for their contributions in building the foundation we have today, and I am excited to work closely with Jonathan as we continue to build long-term value across the pipeline and become a clinical-stage company.”
Mr. Montagu has served as an OMass Board Director since 2019 and brings extensive experience as a trained chemist and biotechnology executive, with deep expertise across immunology, smallmolecule drug discovery platforms and leadership of private biotech companies. Prior to cofounding HotSpot, Mr. Montagu served as Chief Business Officer at Nimbus Therapeutics, where he helped establish the company's drug discovery platform and pipeline. He also served as Vice President, Business Development at Concert Pharmaceuticals, where he played a leading role in executing the company’s $1 billion multi-product transaction with GSK.
“I want to thank Jim and Birgitte for their many valuable contributions to OMass as the company has grown and successfully progressed multiple programs to late-stage preclinical development,” said Mr. Montagu. “OMass has built a differentiated pipeline and platform, and with several important milestones ahead, including the planned entry into the clinic, I look forward to working closely with Ros and the leadership team to help guide the company through its next phase of growth.”
For further information, please contact:
OMass Therapeutics
Rosamond Deegan, Chief Executive Officer
Email: ros.deegan@omass.com
THRUST
Renee Leck
Email: renee@thrustsc.com
About OMass Therapeutics
OMass Therapeutics is a biotechnology company discovering medicines against highly validated target ecosystems, such as membrane proteins or intracellular complexes.
OdyssION™, OMass’ unique drug discovery platform, comprises next-generation native mass spectrometry with novel biochemistry techniques and custom chemistry to interrogate not just a drug target, but also the interaction of the target with its native ecosystem, separate from the confounding complexity of the cell. This unique approach results in cell-system fidelity with cell-free precision.
OMass is advancing a pipeline of small molecule therapeutics in rare diseases and immunological conditions. Its lead program is a best-in-class MC2 (melanocortin-2) receptor antagonist for the treatment of Congenital Adrenal Hyperplasia (CAH) and ACTH-dependent Cushing syndrome. The focus of the program has been to increase receptor residency time to make OMass’ antagonists resistant to competition by the endogenous ligand, thereby avoiding loss of efficacy in the face of rising adrenocorticotropic hormone (ACTH) levels due to reductions in glucocorticoid supplementation for CAH or progression of Cushing’s Syndrome.
Headquartered in Oxford, UK, OMass has raised over $160M (£129M) from a top-tier international investor syndicate including Syncona, Oxford Science Enterprises, GV, Northpond Ventures, Sanofi Ventures and British Patient Capital.
To learn more, please visit www.omass.com. Follow us on LinkedIn.
NodThera Announces Presentation at the 44th Annual J.P. Morgan Healthcare Conference
Boston, MA, January 6, 2026 – NodThera, a clinical-stage biotechnology company pioneering a paradigm shift in the treatment of chronic inflammatory diseases through selective NLRP3 inhibition, today announced that Chris Guiffre, J.D., M.B.A., Chief Financial Officer of NodThera, will present at the 44th Annual J.P. Morgan Healthcare Conference being held January 12-15, 2026, in San Francisco, CA.
Details of the presentation are as follows:
44th Annual J.P. Morgan Healthcare Conference
Date: Wednesday, January 14, 2026
Time: 8:00 AM PT / 11:00 AM ET
Location: The Westin St. Francis Hotel, San Francisco, CA
About NLRP3
NLRP3 activation mediates the release of proinflammatory cytokines, including IL-6, IL-18 and IL-1β, driving chronic inflammation in cardiometabolic, neurological and other diseases. NLRP3 Inhibition offers an alternative approach to therapeutic blockade of individual cytokines IL-6, IL-18 or IL-1β.
About NodThera
NodThera, a US and UK based biotech developing best-in-class NLRP3 inhibitors, pioneering a paradigm shift in the treatment of diseases driven by chronic inflammation. NodThera has led the science for a decade; and has built an advanced pipeline of multiple clinical-stage small-molecule NLRP3 inhibitors. Compelling first-in-class clinical data, including in patients with cardiometabolic and neurological diseases, is fuelling rapid clinical development towards multiple approvals.
NodThera is backed by top-tier investors including 5AM Ventures, Blue Owl Capital, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners.
NodThera is headquartered in Boston, Massachusetts, with an R&D base in the UK. Learn more at www.nodthera.com or follow the Company on LinkedIn.
Investors and Media
Argot Partners
nodthera@argotpartners.com
Funds to support ongoing Phase 1 trial of MNA-001 and validation of translational biomarkers for TREM2 function in Alzheimer’s
Work will inform Phase 2 trials assessing the potential of MNA-001 to slow pathology progression and functional decline
Validates Muna’s differentiated all-in-human discovery and validation approach to address urgent unmet need in Alzheimer’s disease
COPENHAGEN, Denmark and BOSTON, U.S., January 8, 2025 – Muna Therapeutics, a clinical-stage biotechnology company focused on developing innovative therapeutics for neurodegenerative diseases, today announced it has been awarded a $1 million research grant from the Alzheimer’s Association to support its ongoing Phase 1 trial of MNA-001—a novel potent, selective and orally administered small molecule, in healthy young and elderly subjects—and its work to identify and validate translational biomarkers of the function and activation of triggering receptor expressed on myeloid cells 2 (TREM2) in the context of Alzheimer’s disease.
These efforts will identify and validate critical tools to monitor MNA-001’s pharmacodynamic impact on TREM2 signaling and microglial function under both healthy and pathological conditions. The resulting insights are essential to inform upcoming Phase 2 trials assessing if TREM2 agonism can slow Alzheimer’s pathology progression and functional decline by enhancing the natural clearance of misfolded proteins, normalizing microglial function and reducing neuroinflammation.
"We are grateful to the Alzheimer’s Association for this award, which will accelerate clinical evaluation of MNA-001 and validation of critical biomarkers of TREM2 agonism," said Rita Balice-Gordon, Ph.D., Chief Executive Officer of Muna Therapeutics. "The grant supports our strategy to shift the treatment paradigm from clearing pathology to bolstering the brain's innate protective mechanisms. By recalibrating microglial activity, we aim to interrupt the damaging feedback loop of neuroinflammation and provide a potentially transformative approach to treating early Alzheimer’s."
This award underscores Muna’s growing momentum and its leadership in the discovery of resilience-based therapeutics to slow or stop devastating diseases like Alzheimer’s and to improve the quality of life for patients and their loved ones.
About Muna Therapeutics
Muna Therapeutics is pioneering a new era of drug discovery for neurodegenerative diseases by focusing on enhancing resilience to the effects of misfolded protein pathology to protect brain functions like cognition. Muna’s all-in-human discovery engine identifies new therapeutic targets that can enhance the brain’s innate protective mechanisms. Muna—which means ‘to remember’ in Old Norse—is developing a portfolio of therapeutics to slow or stop devastating
diseases like Alzheimer’s and other neurodegenerative disorders. For more information, visit www.munatherapeutics.com. Follow Muna on LinkedIn.
Media Contact:
Lia Dangelico
Deerfield Group
Email: lia.dangelico@deerfieldgroup.com

First dose-expansion patient successfully received SB-007
SB-007 addresses the root cause of Stargardt disease with the potential to treat patients across all ABCA4 mutations
SB-007 granted FDA Fast Track designation, following previously received Orphan Drug designation in US and Europe
BARCELONA, January 8, 2026 – SpliceBio, a clinical-stage genetic medicines company pioneering protein splicing to address diseases caused by mutations in large genes, today announced that the first patient has been dosed in the Part B dose-expansion portion of the Phase 1/2 ASTRA clinical trial of SB-007, a dual adeno-associated viral (AAV) vector gene therapy for the treatment of Stargardt disease.
Part B will evaluate two dose levels of SB-007 versus an untreated control in patients with the rare inherited retinal disease. Stargardt disease, which has no approved treatments, is caused by biallelic mutations in the ABCA4 gene leading to a progressive deterioration of central vision and ultimately blindness. SB-007 is designed to address the underlying genetic cause by restoring expression of a functional, full-length ABCA4 protein in the retina, with the potential to treat patients across all ABCA4 mutations.
“Gene therapy has transformative promise in ophthalmology, but its application has been limited by the inability of single-AAV vectors to accommodate large, complex genes such as ABCA4. SB- 007 is a dual-AAV vector that can harness our protein splicing platform and is designed to reconstitute the full-length therapeutic ABCA4 protein,” said Aniz Girach, M.D., Chief Medical Officer of SpliceBio. “Starting the dose-expansion phase of the ASTRA trial is a key milestone for our program, and we look forward to continuing this progress with the support of the trial’s participants and investigators.”
“The use of two viral vectors that recombine once inside retinal cells is a unique approach to restoring the large gene needed in Stargardt disease, and dual vectors might have implications for treating other retinal degenerations,” commented Robert MacLaren, M.D., Ph.D., Professor of Ophthalmology, University of Oxford and National Health Service Gene and Cell Therapy Research Lead, Oxford University Hospitals NHS Foundation Trust. “This unique gene therapy modality has the potential to slow or even halt progression of this debilitating disease, which is the most common cause of inherited blindness in children. We are delighted to have treated the first patient here in Oxford, in the critical second phase of the trial. ”
SpliceBio’s proprietary platform technology leverages engineered inteins to enable protein trans- splicing. This allows the ABCA4 gene to be split into two transgenes that are delivered using dual AAV vectors. Once inside the target cells in the retina, the transgenes are expressed and undergo protein trans-splicing to reconstitute the full-length native ABCA4 protein.
“The ASTRA study is a critical step toward expanding our understanding of Stargardt disease and evaluating the potential of SB-007,” added Mariya Moosajee, M.B.B.S., Ph.D., Professor of Molecular Ophthalmology, University College London Institute of Ophthalmology, and Consultant Ophthalmologist and Head of the Genetics Service, Moorfields Eye Hospital in London. “The clinical insights generated through this study have the potential to advance medicine and, critically, bring new hope to patients and families living with this condition. We are proud to have included the first participant in this groundbreaking trial.”
ASTRA is a multicenter, global clinical trial designed to evaluate the safety, tolerability and efficacy of SB-007. SpliceBio expects to enroll approximately 57 patients aged 12 to 65 with Stargardt disease into Part B. Part A of the study evaluated three dose levels of subretinal SB- 007 in an open-label, dose-escalation design. Part B is randomized, controlled and masked, and will evaluate two dose levels of subretinal SB-007 compared to an untreated control group, with a follow-up of 96 weeks. The primary endpoint is safety and tolerability, assessed by the incidence and severity of ocular and non-ocular adverse events. Secondary endpoints comprise multiple efficacy measures. For more information on the trial, visit ClinicalTrials.gov [NCT06942572].
The U.S. Food and Drug Administration (FDA) recently granted Fast Track designation to SB- 007, a process designed to facilitate the development and expedite the review of drugs that treat serious conditions and fill an unmet medical need. With this designation, SpliceBio is eligible for more frequent communication with the FDA, as well as the potential for expedited regulatory review. SpliceBio is also running the POLARIS study, a natural history study in patients with Stargardt disease.
Dr. MacLaren’s work to develop gene therapies for retinal conditions is supported by the National Institute for Health and Care Research (NIHR) Oxford Biomedical Research Centre.
About Stargardt Disease
Stargardt disease is a rare inherited retinal disease (IRD) that causes progressive vision loss and blindness. It affects an estimated 1 in 8,000 to 10,000 children and adults worldwide. The disease is caused by mutations in the ABCA4 gene, which result in the accumulation of toxic vitamin A byproducts that damage photoreceptor cells in the central region of the retina, known as the macula. Central vision loss is a hallmark of Stargardt disease, and the age of onset is variable, including early-onset forms in children and adolescents as well as late-onset forms in older adults. The large size of the ABCA4 gene exceeds the packaging capacity of standard single AAV vectors and has historically limited traditional gene therapy approaches. There are currently no approved treatments.
About SB-007
SB-007 is an investigational dual adeno-associated virus (AAV) gene therapy for Stargardt disease. It is designed to restore expression of a functional, full-length ABCA4 protein in the retina, with the potential to treat patients with all ABCA4 mutations. SpliceBio’s proprietary protein splicing intein platform uses two AAV serotype 8 (AAV8) vectors to overcome the size limitations of conventional AAVs, reconstituting biologically active ABCA4 through protein trans-splicing in target photoreceptor cells. In preclinical models, SB-007 demonstrated robust pharmacological activity, durable retinal expression and a favorable safety profile. SpliceBio has received Orphan Drug designation for SB-007 from the FDA and the European Commission.
About SpliceBio
SpliceBio is a clinical-stage genetic medicines company pioneering protein splicing to address diseases caused by mutations in large genes. Our lead program, SB-007, is a gene therapy designed to target the root cause of Stargardt disease, an inherited retinal disorder that causes progressive vision loss and blindness. SpliceBio is currently enrolling participants in ASTRA, a Phase 1/2 clinical trial of SB-007, and POLARIS, a natural history study in patients with the disease. The SpliceBio platform combines advanced intein, protein splicing and protein engineering technologies, and supports a pipeline of gene therapy programs in ophthalmology and neurology. For additional information, visit www.splice.bio and follow us on LinkedIn and X.
Media Contact:
Amanda Lazaro, 1AB
amanda@1abmedia.com

Financing co-led by Sanofi Ventures and Janus Henderson Investors, with participation from new investors Deep Track Capital, EcoR1 Capital, Logos Capital, Balyasny Asset Management L.P., Woodline Partners LP and all existing investors
Proceeds to fund clinical development of DIAG723, a first-in-class, disease-modifying therapy designed to correct the root cause of hereditary hemorrhagic telangiectasia and pulmonary arterial hypertension, and advance additional clustering antibody drug candidates
DIAG723 is expected to enter the clinic in first half of 2026
Watertown, Mass., January 8, 2026 – Diagonal Therapeutics, a biotechnology company developing disease-modifying clustering antibodies that correct dysregulated signaling in severe genetic diseases, today announced the closing of an oversubscribed $125 million Series B financing co-led by Sanofi Ventures and Janus Henderson Investors, with participation from both existing and new investors. New investors include Deep Track Capital, EcoR1 Capital, Logos Capital, Balyasny Asset Management L.P. and Woodline Partners LP. Existing investors include Atlas Venture, BVF Partners, Lightspeed Venture Partners, RA Capital Management, Frazier Life Sciences, Viking Global Investors, RV Invest, Velosity Capital, Biovision Ventures, and Checkpoint Capital. Paulina Hill, Ph.D., Partner at Sanofi Ventures, will join Diagonal’s Board of Directors in conjunction with the financing.
Proceeds will support advancement of DIAG723, a first-in-class, disease-modifying clustering antibody designed to correct the underlying cause of hereditary hemorrhagic telangiectasia (HHT) and pulmonary arterial hypertension (PAH), conditions that collectively impact hundreds of thousands of people around the world. Diagonal expects to initiate its first-in-human trial for DIAG723 in HHT patients during the first half of 2026.
“DIAG723 has exhibited robust disease-modifying activity in several preclinical models of HHT and PAH, demonstrating its ability to prevent and reverse disease pathology and restore normal signaling in cells derived from patients,” said Alex Lugovskoy, Ph.D., Founder and CEO of Diagonal Therapeutics. “We are deeply grateful to our new and existing investors for their support as we advance DIAG723 into the clinic, with the goal of delivering a life-changing therapy for patients in need of effective treatment options. This financing also enables us to advance our pipeline of additional clustering antibody therapeutics.”
“Diagonal’s unique clustering antibody approach has the potential to address the root cause of genetic diseases that originate from impaired receptor signaling,” said Paulina Hill, Ph.D., Partner at Sanofi Ventures. “We are excited to support this innovative company led by an experienced team with a strong track record of operational excellence, deep commitment to improving patients’ lives, and bold vision for transforming care in severe genetic diseases.”
###
About Hereditary Hemorrhagic Telangiectasia (HHT)
HHT is a rare disease that affects more than 330,000 people in the U.S. and EU, and for which
there are currently no approved therapies. In HHT, loss of function point mutations in members
of the TGF-ß receptor superfamily complex create abnormal blood vessels that are fragile and
susceptible to rupture and bleeding. These bleeding events drastically reduce quality of life,
result in emergency room visits and hospitalizations, and lead to chronic anemia, necessitating
frequent iron infusions or red blood cell transfusions in many patients. Solid organ arteriovenous
malformations, if left untreated, are at risk of rupturing, resulting in lung and brain hemorrhage,
stroke, heart failure, or death.
About Pulmonary Arterial Hypertension (PAH)
PAH is a rare, progressive, life-threatening disorder that impacts an estimated 200,000 patients
globally. Current PAH treatments can manage symptoms and slow disease progression but
there have been reports of the formation of telangiectasia, epistaxis and pulmonary shunting
with recent modulators of the pro-proliferative pathway. In PAH, loss of function mutations in
BMPR2 or BMPRII downregulation leads to impaired ALK1 signaling that causes vascular
hyperproliferation, narrowing the pulmonary arterial walls and resulting in elevated pulmonary
arterial pressure and ultimately death due to right ventricular failure.
About DIAG723
DIAG723 is a clustering antibody designed specifically to address the root cause of HHT and
PAH, in which dysregulated ALK1 signaling in endothelial cells drives the formation of fragile
arteriovenous malformations or vascular hyperproliferation, respectively. DIAG723 restores
signaling lost due to mutations that impair receptor function. In multiple HHT preclinical studies,
DIAG723 prevented and reversed arteriovenous malformations and anemia – a hallmark of HHT
that can cause a host of bleeding-related complications in various organs. DIAG723 was also
found to restore signaling in multiple HHT patient donor samples. In preclinical models of PAH,
DIAG723 prevented disease development and cardiac remodeling, and improved
hemodynamics. DIAG723 also restored normal signaling in pulmonary microvascular
endothelial cells derived from multiple PAH donors. DIAG723 has received orphan drug
designation from the US FDA and the EMA for the treatment of HHT.
About Diagonal Therapeutics
Diagonal Therapeutics is a biotech company advancing novel disease-modifying clustering
antibodies that repair dysregulated signaling implicated in a range of illnesses. The Company's
DIAGONAL Product Engine combines proprietary computational and experimental techniques
to overcome historical challenges associated with antibody drug discovery and efficiently deliver
optimized therapeutic assets. Diagonal's pipeline comprises clustering antibodies designed to
selectively address the underlying cause of disease across hematology, hepatology, and nephrology, offering the potential to deliver life-changing therapies for patients. For more
information, please visit www.diagonaltx.com.
Media Contact
media@diagonaltx.com
BARCELONA, January 06, 2026 – SpliceBio, a clinical-stage genetic medicines company pioneering protein splicing to address diseases caused by mutations in large genes, today announced the appointment of Don Munoz as Chief Financial Officer. Mr. Munoz, who is based in Boston, is a seasoned biotechnology finance leader with deep experience in financial strategy, operations and corporate development, complemented by a strong background in healthcare investment banking.
“As we advance our lead program, SB-007 for Stargardt disease, through the clinic and continue to expand SpliceBio’s pipeline, strengthening our financial leadership is a critical priority,” said Miquel Vila-Perelló, Ph.D., CEO and Co-Founder of SpliceBio. “Don brings exceptional experience in guiding biotechnology companies through periods of clinical execution, capital formation and strategic growth. His leadership will be instrumental as we enter our next stages of development and position SpliceBio for long-term value creation.”
Mr. Munoz most recently served as CFO of Aurion Biotech, where he helped lead the ocular cell therapy company’s acquisition by Alcon in March 2025. Previously, as CFO at NuCana, he led capital formation, partnering and financing strategy, including its $114 million initial public offering and $80 million follow-on offering. Mr. Munoz also served as Group CFO of Noxxon Pharma. Earlier in his career, Mr. Munoz held senior leadership roles in healthcare investment banking, including Managing Director and Head of Medical Technology Investment Banking at both Cowen & Company and Leerink Partners, as well as leadership positions at Deutsche Bank and its predecessor, Alex. Brown & Sons. Mr. Munoz holds a BA from Dartmouth College and an MBA from Columbia Business School.
“SpliceBio has built a differentiated genetic medicines platform with the potential to address diseases that remain underserved by current approaches,” said Mr. Munoz. “With impressive clinical progress and a strong leadership team in place, the company is entering an exciting phase of development. I am thrilled to join SpliceBio and to help support the company’s continued growth and long-term success for patients.”
About SpliceBio
SpliceBio is a clinical-stage genetic medicines company pioneering protein splicing to address diseases caused by mutations in large genes. Our lead program, SB-007, is a gene therapy designed to target the root cause of Stargardt disease, an inherited retinal disorder that causes progressive vision loss and blindness. SpliceBio is currently enrolling participants in ASTRA, a Phase 1/2 clinical trial of SB-007, and POLARIS, a natural history study in patients with the disease. The SpliceBio platform combines advanced intein, protein splicing and protein engineering technologies, and supports a pipeline of gene therapy programs in ophthalmology and neurology. For additional information, visit www.splice.bio and follow us on LinkedIn and X.
Media Contact:
Amanda Lazaro, 1AB
amanda@1abmedia.com
- Seasoned physician-executive with global biopharma leadership experience joins Board at a pivotal stage of clinical and corporate development
Boston, MA, December 16, 2025 – NodThera, a clinical-stage biotechnology company pioneering a paradigm shift in the treatment of chronic inflammatory diseases through selective NLRP3 inhibition, today announced the appointment of Geoff McDonough, M.D., as an Independent Board Member.
Dr. McDonough brings more than two decades of leadership experience spanning R&D, commercial strategy, and global company building. He most recently served as President and Chief Executive Officer of Generation Bio (NASDAQ: GBIO). Dr. McDonough previously served as President and CEO of Sobi, overseeing substantial portfolio expansion and international growth. Earlier in his career, Dr. McDonough spent a decade at Genzyme, holding senior roles including President of Genzyme Europe and Senior Vice President & General Manager of the global Lysosomal Storage Diseases business. He trained in internal medicine and pediatrics at Massachusetts General Hospital and Boston Children’s Hospital, and earned his M.D. from Harvard Medical School.
“We are excited to welcome Geoff to NodThera’s Board,” said Don Nicholson, Ph.D., Executive Chair of the Board. “His breadth of global biopharma leadership experience and his proven strategic insight will be invaluable as we continue to advance our cardiometabolic Phase 2 clinical trials and, ultimately, deliver valuable medicines patients with chronic inflammatory diseases.
“It is a privilege to join NodThera’s Board as the Company approaches two critical Phase 2 readouts next year for NT-0796, a unique brain-penetrant, oral small molecule NLRP3 inhibitor,” said Dr. McDonough. “I look forward to working with the Board of Directors and management team to change what’s possible for millions of patients and families suffering from cardiometabolic disease.”
About NLRP3
NLRP3 activation mediates the release of proinflammatory cytokines, including IL-6, IL-18 and IL-1β, driving chronic inflammation in cardiometabolic, neurological and other diseases. NLRP3 Inhibition offers an alternative approach to therapeutic blockade of individual cytokines IL-6, IL-18 or IL-1β.
About NodThera
NodThera, a US and UK based biotech developing best-in-class NLRP3 inhibitors, pioneering a paradigm shift in the treatment of diseases driven by chronic inflammation. NodThera has led the science for a decade; and has built an advanced pipeline of multiple clinical-stage small-molecule NLRP3 inhibitors. Compelling first-in-class clinical data, including in patients with cardiometabolic and neurological diseases, is fuelling rapid clinical development towards multiple approvals.
NodThera is backed by top-tier investors including 5AM Ventures, Blue Owl Capital, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners.
NodThera is headquartered in Boston, Massachusetts, with R&D base in the UK. Learn more at www.nodthera.com or follow the Company on LinkedIn.
Investors and Media
Argot Partners
nodthera@argotpartners.com

Co-led by SemperVirens Venture Capital and Next Ventures, with participation by Sanofi Ventures, Lux Capital, Civilization Ventures, How Women Invest and other notable investors
SAN FRANCISCO, DECEMBER 2, 2025 – Trial Library, an AI-enabled platform expanding access to clinical trials as a care option, announced a $10 million Series A funding round co-led by SemperVirens Venture Capital and Next Ventures, with participation from Sanofi Ventures, Lux Capital, Civilization Ventures, How Women Invest, Overwater Ventures and others. This investment, which brings total funding to date of $15 million, accelerates Trial Library’s mission to make clinical research a core part of standard of care – bridging access gaps for patients, empowering healthcare providers and aligning payers and biopharmaceutical sponsors around a more efficient and equitable research ecosystem.
“As a physician-scientist, I witnessed, first-hand, the significant impact clinical trials have on improved healthcare outcomes. At Trial Library, we’re building the connective tissue that links care delivery and clinical research, ” said Dr. Hala Borno, CEO and founder of Trial Library. “This latest funding will enable us to advance our model where access to innovation is the standard, not the exception.”
Trial Library’s platform unlocks access to clinical trials in community-based clinical settings through AI-enabled provider activation, compliant eligible patient identification and longitudinal navigation throughout the trial lifecycle. Trial Library currently operates across 320+ clinics and 1,500+ providers nationwide and is rapidly expanding its network alongside growing partnerships with payers such as self-insured plans and health plans.
“Trial Library is the first technology company to unlock the payer market to effectively broaden the pool of patients who are considered for clinical trials, ” said Allison Baum Gates, general partner of SemperVirens Venture Capital. “We’re excited to back a platform that aligns incentives and unites patients, providers, payers, and life-science partners around shared cost-savings and patient outcomes. ”
Trial Library has partnered with leading global biopharmaceutical companies to accelerate R&D and enrollment. Ventures. “Trial Library is a uniquely end-to-end platform, building the AI infrastructure that is accelerating access to the next generation of precision medicine, ” said Cris De Luca, partner at Sanofi Ventures.
Since launching in 2022, Trial Library has demonstrated significant impact through strategic partnerships with leading healthcare organizations. The company's work with The Oncology Institute generated 642 patient referrals in one year, while partnerships with American Oncology Network have identified tens of eligible patients for solid tumor trials in just the first quarter of implementation. Trial Library’s platform has also expanded its collaboration with Johnson & Johnson Innovative Medicine to support oncology clinical development and medical affairs programs, and in 2025 the company announced a collaboration with Texas Oncology to streamline clinical trial access through personalized patient navigation
“Trial Library’s approach aligns with the growing shift toward decentralized, community-based research, ” said Lance Armstrong, managing partner at Next Ventures. They’re proving that AI and human navigation together can bring precision medicine to every corner of healthcare. ”
Trial Library is committed to diversity and equity among patient populations, including a focus on underserved populations. This includes 69% of patient referrals being non-white, 29% on Medicaid and 44% requiring transportation support. This effort addresses critical gaps in clinical trial participation while reducing administrative burden on providers and transforming how community oncology practices engage with clinical research.
About Trial Library
Trial Library is an AI-platform that accelerates access to precision medicine. In collaboration with biopharmaceutical manufacturers, payers and health systems, Trial Library enables the delivery of clinical trials as a care option, advancing access to precision medicine, improving oncology outcomes and reducing the total cost of care. Backed by some of the leading venture capital firms in healthcare, Trial Library currently operates across 320+ clinics and 1,500+ providers nationwide. Learn more at http://www.triallibrary.com
Media Contact
Lizi Sprague, Songue PR
lizi@songuepr.com
Company Raised $112 Million to Date and is on Track to Generate Initial Clinical Data by Year End
BURLINGTON, MA – November 18, 2025 -- Lifordi Immunotherapeutics, Inc., a clinical-stage biotech company developing antibody-drug conjugates (ADCs) for the treatment of autoimmune and inflammatory disorders, today announced a strategic investment from Sanofi Ventures, the venture arm of global healthcare leader Sanofi, and funding from existing investors ARCH Ventures, 5AM Ventures, and Atlas Venture. The new funds bring the total raised to $112 million and support an ongoing Phase 1 study in Rheumatoid Arthritis (RA) designed to evaluate LFD-200, an ADC delivering a potent glucocorticoid (GC) directly to immune cells. It also provides for Chemistry Manufacturing and Controls (CMC) preparations to ensure that Phase 2 clinical supply is available without incurring an unnecessary delay. As part of the investment, Christopher Gagliardi, Ph.D., Principal at Sanofi Ventures, will join as an observer on Lifordi’s Board of Directors.
“Sanofi has demonstrated a strong commitment to invest in new treatments for autoimmune and inflammatory diseases, and we are fortunate to have this support from Chris and the Sanofi Ventures team alongside our current investors to advance LFD-200 and our targeted ADC delivery pipeline for immune-mediated conditions, ” said Arthur Tzianabos, Ph.D., President & Chief Executive Officer. “Enrollment and dosing in our Phase 1 study of LFD-200 in RA is progressing as planned, and we look forward to sharing initial data from healthy participants in the coming months.”
“We continue to search for new approaches to improve the treatment of autoimmune and inflammatory diseases and were intrigued by Lifordi’s targeted ADC approach to deliver glucocorticoids without toxicity,” said Christopher Gagliardi, Ph.D. “Once we met the team and did our due diligence on the proof-of-concept data in multiple animal models of autoimmune disease together with extensive nonclinical studies, we made the decision to invest in Lifordi now. This enables Sanofi to share our expertise and experiences to help guide LFD-200 through clinical studies and support pipeline development using this approach to deliver other drug payloads, such as ASOs or siRNAs.”
Lifordi’s Phase 1 clinical trial is currently enrolling and dosing healthy participants. The study is designed to evaluate the safety and efficacy of LFD-200, and initial healthy participant data will include both safety and pharmacodynamic measures. Following single and multiple ascending dosing (SAD/MAD studies), the Company plans to evaluate LFD-200 in patients with moderate to severe rheumatoid arthritis. Lifordi recently presented non-clinical data at the American College of Rheumatology (ACR) 2025 meeting showing that clinically relevant doses of LFD-200 given subcutaneously every 7 days for 13 weeks maintained glucocorticoid exposure in immune cells without evidence of systemic toxicity. By harnessing the efficacy of GCs while limiting the toxicity, LFD-200 has the potential to solve the problem that has limited the broad and long-term use of GCs for the past 75 years.
About Lifordi
Lifordi Immunotherapeutics, Inc. is a clinical-stage biotechnology company leading the way by leveraging the success of antibody-drug conjugates (ADCs) to develop treatments for autoimmune and inflammatory disorders. The Company’s lead ADC, LFD-200, is in a Phase 1 clinical trial. Preclinical studies demonstrated efficacy in multiple disease models by targeting myeloid and lymphoid cells using a highly internalized cell surface membrane protein (VISTA). Lifordi has also applied its novel drug delivery platform to other diverse payloads, such as small molecules, antisense oligonucleotides (ASOs) and siRNA. As experienced drug developers in immunology and inflammatory diseases, together with expert clinical advisors, a strong partnering track record, and funding from ARCH Venture Partners, 5AM Ventures, and Atlas Venture, Lifordi is committed to changing how immune and inflammatory diseases are treated. For more information, please visit www.lifordi.com.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi, investing globally in early-stage biotech and digital health companies aligned with Sanofi’s mission to chase the miracles of science. Areas of focus include immunology, oncology, rare diseases, vaccines, and digital innovation.
Contact
Theresa McNeely
Chief Communications Officer
tmcneely@lifordi.com
(508) 523-9511
MNA-001 has best-in-class potential based on preclinical data demonstrating ability to significantly reduce neurotoxic amyloid burden and reprogram microglia to protective states
Phase 1 study initiated to assess safety, tolerability, pharmacokinetics and pharmacodynamic effects of MNA-001 on biomarkers of TREM2 target engagement and activation, with topline data expected mid 2026
Achievement marks Muna’s evolution into a clinical-stage company
COPENHAGEN, Denmark, November 10, 2025 – Muna Therapeutics, a clinical stage biotechnology company focused on developing innovative therapeutics for neurodegenerative diseases, today announced that the first subjects have been dosed in a Phase 1 single-ascending dose/multiple-ascending dose study of lead asset MNA-001 in healthy adult participants. MNA-001 is a novel potent, selective and orally administered small molecule “triggering receptor expressed on myeloid cells 2” (TREM2) agonist with best-in-class potential for the treatment of early Alzheimer’s and other neurodegenerative diseases.
“We are delighted to have initiated clinical development of MNA-001. Our goal is to fundamentally shift the focus in Alzheimer’s disease treatment from clearing pathology to bolstering the brain's innate protective mechanisms,” said Rita Balice-Gordon, Ph.D., Muna’s Chief Executive Officer. “This milestone demonstrates our progress toward advancing disease-modifying therapeutics that mitigate key drivers of neurodegeneration and functional impairment to improve disease outcomes.”
MNA-001 is designed to activate TREM2, a critical regulator of microglia, which are brain cells with immune and other functions. By boosting TREM2 signaling, MNA-001 enhances protective microglial responses to pathology, promoting the phagocytosis of toxic misfolded proteins and cell debris to slow or stop the progression of neurodegeneration. MNA-001’s best-in-class potential is supported by a strong preclinical data package demonstrating the significant reduction of amyloid pathology and reprogramming of disease-associated microglia to homeostatic, protective states in animal models of Alzheimer’s pathology. The molecule was shown to be safe and well tolerated in preclinical toxicology studies.
The primary objective of Muna’s Phase 1 randomized, double-blind, placebo-controlled study is to evaluate the safety and tolerability of MNA-001 in healthy adult and elderly subjects. The study will also assess pharmacokinetics and the pharmacodynamic effects of MNA-001 on biomarkers of TREM2 engagement and activation in plasma and CSF. It also will leverage Muna’s novel biomarker discoveries to provide early insights into MNA-001’s activity in humans, with topline data expected in late summer, 2026.
“We believe that MNA-001 has the potential to treat disease and improve quality of life for Alzheimer’s patients and their loved ones,” said Donald Nicholson, Ph.D., Chair of Muna’s Board of Directors. “We intend for this to be the first of several innovative approaches to enhance the brain’s resilience to pathological drivers of neurodegeneration.”
About Muna Therapeutics
Muna Therapeutics is pioneering a new era of drug discovery for neurodegenerative diseases by focusing on enhancing resilience to the effects of misfolded protein pathology to protect brain functions like cognition. Muna’s all-in-human discovery engine identifies new therapeutic targets that can enhance the brain’s innate protective mechanisms. Muna—which means ‘to remember’ in Old Norse—is developing a portfolio of therapeutics to slow or stop devastating diseases like Alzheimer’s and other neurodegenerative disorders. For more information, visit www.munatherapeutics.com. Follow Muna on LinkedIn.
###
Media Contact:
Lia Dangelico
Deerfield Group
- $32 million new equity injection adds to previously announced $59 million
- New investors Tencent and BGF joined by all existing major shareholders
- Funds will advance pipeline of novel TCR-CD3 bispecifics in cancer and autoimmune disease
13 November 2025; Cambridge, England – T-Therapeutics, a biotechnology company developing nextgeneration T cell receptor (TCR) therapeutics for cancer and autoimmune disease, today announces the successful expansion of its Series A financing, raising a further $32 million. Following the initial $59 million raised, this brings the Series A total to date to $91 million.
New investors Tencent and BGF joined the Series A syndicate, alongside all existing major shareholders Sofinnova Partners, F-Prime, Digitalis Ventures, Cambridge Innovation Capital, Sanofi Ventures and the University of Cambridge Venture Fund.
T-Therapeutics will use the additional proceeds to drive its pipeline of first-in-class TCR-CD3 bispecifics across oncology and autoimmune diseases towards the clinic, including the further exploration of new therapeutic strategies such as T cell subset depletion. T-Therapeutics’ lead asset in oncology exploits a pan-tumour driver target, applicable across multiple different solid tumour types. Its lead immunology programme is a pan-autoimmune bispecific designed for precision immune reset, achieved by the selective depletion of pathogenic immune cells.
Theodora Harold, Chief Executive Officer of T-Therapeutics, said: “Our transformative medicines tackle upstream disease-drivers that can have pan-indication impact. We are delighted to have significantly added to our Series A financing, which we see as a strong validation of both our technology and our progress to date. I would like to thank Tencent and BGF for their belief in our potential, as well as all our existing investors for their continued support.”
T-Therapeutics’ TCR platform, OpTiMus®, can generate an almost unlimited repertoire of high specificity, fully human TCRs, enabling access to validated, but previously undruggable, intracellular targets. The Company also leverages its proprietary next-generation CD3 T cell engagers (TCEs), which have been engineered for high potency, superior safety and favourable pharmacokinetics. OpTiMus-derived TCRs are combined with the proprietary TCEs to form first-in-class bispecific drug candidates. T-Therapeutics’ pipeline is focused on upstream disease-drivers with pan-indication potential to deliver significant clinical benefit for patients.
Graziano Seghezzi, Managing Partner at Sofinnova Partners, said: “The existing investors co-founded TTherapeutics to push the boundaries of bispecific technology. This additional capital enables us to expand into T cell subset depletion, one of the most exciting areas in immunology, while continuing to advance oncology programmes. We are now ideally positioned to address both cancer and autoimmune disease, two broad disease areas with critical unmet medical needs, with a platform that unlocks targets previously considered undruggable.”
Luke Rajah, Partner at BGF, added: "This is a leadership team with an outstanding track record of building successful drug discovery businesses and translating science into medicines. Backed by a syndicate of world-class life sciences investors, T-Therapeutics is uniquely positioned to unlock previously undruggable targets with its first-in-class bispecifics. We are proud to support the team and help catalyse their programmes towards the clinic.”
Contact Us
T-Therapeutics
Theodora Harold
info@t-therapeutics.com
ICR Healthcare
Amber Fennell, David Daley, Lucy Featherstone
t-therapeutics@icrhealthcare.com
About T-Therapeutics
T-Therapeutics is a next-generation T cell receptor (TCR) company spun out from the University of Cambridge. The company was created to harness the power of T cell biology, to create safe and effective treatments for cancer and autoimmune disease. T-Therapeutics' experienced team combines worldleading expertise in mouse genome engineering, single cell genomics, biopharmaceutical drug development, machine-learning and structural biology, anchored in a culture of creativity and collaboration. T-Therapeutics is developing ‘optimal’ TCR therapeutics using its proprietary OpTiMus® discovery platform, that provides an almost unlimited source of unique, antigen-specific human TCRs. The company is developing a pipeline of first-in-class drugs that are intended to become transformative medicines, reshaping the clinical landscape for patients with cancer or autoimmune diseases.
The company is backed by blue-chip investors, including Sofinnova Partners, F-Prime, Digitalis Ventures, Cambridge Innovation Capital, Tencent, BGF, Sanofi Ventures and, through Cambridge Enterprise, the University of Cambridge Venture Fund.
Dr Lee is a seasoned biopharmaceutical executive with a proven track record of leading cross-functional teams to successfully develop and commercialize innovative therapies in oncology.
Sydney, Australia and Boston, MA, USA – November 17, 2025 – AdvanCell Pty Ltd (“AdvanCell”), a clinical-stage radiopharmaceutical company developing innovative targeted alpha therapies for cancer, today announced the appointment of Philina Lee, PhD as Chief Executive Officer and member of the Board of Directors. Dr Lee will head AdvanCell’s US expansion, accelerating clinical, manufacturing, and partnership activities to advance lead clinical candidate 212Pb-ADVC001 and the company’s innovative radiopharmaceutical portfolio. She will assume her role from January 1, 2026, and will be based at the company’s new US headquarters near Boston/Cambridge, MA.
"Philina brings a rare combination of global commercial leadership, deep radiopharmaceutical expertise, and an outstanding record of building high growth organizations and high-performance teams,” said Andrew Kay, Chair of AdvanCell. “Her leadership and experience positions AdvanCell to drive its next phase of growth, with the United States now a major strategic focus as the company advances 212Pb-ADVC001 and its emerging pipeline of targeted alpha therapies in the world’s largest pharmaceutical market."
Dr Lee joins AdvanCell from Blueprint Medicines Corporation, recently acquired by Sanofi for approximately US$9.5 billion. At Blueprint, she served as Chief Commercial Officer and was instrumental in driving the company’s strategy and building commercial infrastructure in oncology and mast cell disorders. She also served on the Board of Directors of radiopharmaceutical company Fusion Pharmaceuticals Inc. and held senior roles in the US at alpha therapy pioneer Algeta ASA, where she contributed to building the organization that successfully launched the first and only approved alpha therapy Xofigo®* (radium-223 dichloride) in the US in collaboration with Bayer AG.
Dr Lee earned a B.S. in Biochemistry from the University of Alberta, and a PhD in Cell Biology from the Massachusetts Institute of Technology.
"AdvanCell is pioneering a new class of targeted alpha therapies powered by 212Pb (Lead-212) with tremendous potential to transform cancer treatment and position the company as the leading targeted alpha radiotherapy company,” said Dr Philina Lee. “The clinical data from the Phase 1b part of the TheraPb trial recently presented for 212Pb- ADVC001 in prostate cancer at ESMO are promising, demonstrating an encouraging safety profile and strong therapeutic index, including a 100% overall rate of response in patients with RECIST-measurable lesions. I am excited to accelerate the expansion of our US leadership and operational capabilities, building on AdvanCell’s strong foundation in Australia and advancing our mission to transform care for cancer patients globally."
Dr Lee succeeds founder and long-time CEO Andrew Adamovich, who will continue as Managing Director, Australia, and remain on the Board of Directors. The Board acknowledges Mr Adamovich’s leadership in growing AdvanCell from concept to a leading clinical-stage, vertically integrated radiopharmaceutical company, with manufacturing in Australia, a growing US presence, and strategic partnerships across isotope supply, R&D, and clinical translation.
"AdvanCell’s growth trajectory is remarkable – driven by our team’s relentless focus on improving outcomes for cancer patients based on innovative science and strong execution," said Andrew Adamovich. "Philina's leadership further strengthens our ability to establish the United States as a global hub while continuing to deliver innovation from Australia and cementing our position as a leader in targeted alpha radiotherapies."
* Xofigo® is a registered trademark of Bayer AG, used under license (approved in the US, EU, China, and other markets worldwide)
ESMO – European Society for Medical Oncology 2025 Congress
About 212Pb-ADVC001
212Pb-ADVC001 is a proprietary and patented PSMA-targeting radioligand with optimized physicochemical properties and labelled with 212Pb, an alpha-emitting payload (radionuclide) with a high dose rate, 10.6 hour half-life and simple decay scheme. 212Pb-ADVC001 is designed to deliver radiation at a cellular level to effectively kill prostate cancer cells while minimizing toxicity.
212Pb-ADVC001 is being evaluated in the TheraPb trial (NCT05720130) – a prospective, open-label Phase 1/2 dose-escalation and expansion study to assess the efficacy and safety of 212Pb-ADVC001 in men with prostate cancer.
The Phase 1b dose escalation (n=22) showed a compelling therapeutic index for 212Pb- ADVC001, demonstrating encouraging safety and promising anti-tumor activity, including no dose-limiting toxicities or treatment-related serious adverse events, 80% PSA50 response at doses ≥ 160 MBq and 100% objective response rate (ORR) in patients with RECIST-measurable lesions, including two complete responses (see results presented at ESMO 2025 here).
Phase 2 utilizes a randomized dose-response design and adaptive dosing elements to evaluate optimal dosing strategies of 212Pb-ADVC001 in PSMA-positive mCRPC and in mHSPC.
About AdvanCell
AdvanCell is a vertically integrated, clinical-stage radiopharmaceutical company dedicated to developing innovative cancer therapies that harness the power of targeted alpha-emitting radionuclides. By leveraging its proprietary Lead-212 platform, advanced and scalable manufacturing capabilities and world-class clinical development capabilities, AdvanCell aims to deliver novel treatments that improve outcomes for patients with cancer globally. For more information, visit www.advancell.com.au and follow us on LinkedIn.
Contacts
Andrew Adamovich, CEO
contact@advancell.com.au,
For media inquiries, please contact:
MEDiSTRAVA (in the UK)
Mark Swallow, Frazer Hall, Sylvie Berrebi
advancell@medistrava.com
+44 (0)20 3928 6700

- Phase 1a/1b study will evaluate NRM-823 as monotherapy and in combination with immune checkpoint inhibition in patients with locally-advanced or metastatic refractory solid tumors
- NRM-823 is a T cell engager against an immuno-active novel target that is homogeneously and commonly expressed in a broad range of solid tumors
Boston, Mass., and New Haven, Conn. – November 5, 2025 – Normunity, a biotechnology company pioneering precision biologics, today announced that the first patient has been dosed in a Phase 1 clinical trial (NCT07182149) for its lead drug candidate, NRM‑823, a first-in-class T cell engager against a novel immuno-active tumor-specific target. The trial is designed to evaluate the safety, tolerability and initial efficacy of NRM‑823 in increasing dose levels as a monotherapy and in combination with immune checkpoint inhibition in patients with locally-advanced or metastatic refractory solid tumors that express the relevant target.
“The initiation of this Phase 1 clinical trial represents an important milestone for Normunity, as we bring a potentially transformational cancer therapy forward to patients and transition to a clinical-stage company,” said Rachel Humphrey, MD, Chief Executive Officer of Normunity. “This study is the initial step utilizing our first in a new class of Immuno-smart targets for our T cell engager, NRM‑823, and we plan to leverage this target in broader therapeutic modalities beyond T cell engagers. We believe that NRM‑823 exemplifies the power of our proprietary discovery platform to uncover new biologic insights specific to both the immune system and tumors, enabling us to translate our science into potential life-changing medicines for cancer patients.”
Preclinical data with NRM-823 has demonstrated potent anti-tumor activity in vitro and in vivo with T cell-mediated cytotoxicity in humanized mouse models of multiple cancers. NRM-823 has also demonstrated a favorable pharmacokinetic and preclinical safety profile, including no observed adverse events in the GLP toxicity study in non-human primates at the (supratherapeutic) highest dose tested.
“Normunity’s design and development of NRM‑823 allows us to harness the body’s own immune system by activating and redirecting T cells to precisely target tumor cells. Our team is particularly encouraged by the ability of NRM-823 to address a previously unrecognized target that appears to support cancer survival across a range of solid tumor indications,” said Melinda Merchant, MD, PhD, Chief Medical Officer of Normunity. “The compelling preclinical efficacy and safety data give us confidence to move into the clinic to evaluate the potential of NRM‑823 for patients with advanced or metastatic solid tumors.”
About Normunity
Normunity is a biotechnology company creating novel anti-cancer therapies focused on untapped biology at the interface of the immune system and the tumor, addressing Immuno-smart targets responsible for tumor-specific immune suppression that are identified with Normunity’s proprietary target discovery platform. The company is using these novel targets to build a pipeline of anti-cancer medicines, including therapeutic antibodies, bispecific antibodies, and payload-carrying biologics. The company’s lead clinical candidate, NRM‑823, is a first-in-class T cell engager that binds a novel, highly tumor-specific target expressed on multiple types of solid tumors but is absent in normal systemic tissues. Normunity is located in Boston, MA, and New Haven, CT. For more information, please visit www.normunity.com and follow us on LinkedIn.
Media Contact
Kathryn Morris, The Yates Network LLC
914-204-6412
Discovery and development of the first systemic therapeutics to deliver self-antigens directly to the thymus to harness the body’s natural mechanism to induce central tolerance
Lead program in Type 1 diabetes is advancing toward the clinic, with pipeline opportunities in a broad range of autoimmune diseases
Company appoints Jason F. Cole, experienced biotech leader and company builder, as Chief Executive Officer
CAMBRIDGE, Mass., October 28, 2025 — Zag Bio™, Inc., a biotechnology company pioneering thymus-targeted medicines, today announced the company’s launch with $80 million in financing, including a recently closed Series A financing. Polaris Partners founded and incubated Zag Bio and co-led the Series A financing with the T1D Fund, with participation from Mission BioCapital, AbbVie Ventures, Lightspeed Ventures, Sanofi Ventures, KdT Ventures, Regeneron Ventures, Boxer Capital and Pear VC. Zag Bio also announced the appointment of biotech industry veteran Jason F. Cole, J.D., as the company’s Chief Executive Officer and Board member.
Zag Bio has discovered a novel approach to deliver tolerizing antigens to the thymus to achieve durable antigen-specific central tolerance to treat autoimmune disease. Zag Bio designs thymus-targeting bifunctional antibodies to deliver antigens to the thymus, where it engages the body’s natural mechanism for T cell tolerance, expanding antigen-specific thymic regulatory cells (Tregs) while depleting antigen-specific T effector cells (Teff). The thymic Tregs then migrate to diseased tissues to exert a broad range of protective effects. Thymic Tregs are epigenetically hardwired to be stable and durable, and are expected to enable an immune reset with long-lasting impact.
The proceeds from the financing will be used to advance Zag Bio’s lead program into the clinic as a tolerogenic therapy to prevent or delay the onset of Type 1 diabetes. In addition, the financing will enable the company to further advance its discovery programs for a range of other autoimmune diseases. Zag Bio’s proprietary technology facilitates modular design of thymus-targeted self-antigens, enabling streamlined development of additional pipeline programs.
“The significant progress by the Zag Bio team coupled with our recent financing enables the company to rapidly advance our lead program to the clinic. Zag Bio is trailblazing an unprecedented therapeutic pathway for medicines to target the thymus, and this positions the company to be an innovator in autoimmune disease treatment with the potential to help millions of patients,” said Alan Crane, Co-Founder and Chairman of Zag Bio and Entrepreneur Partner at Polaris Partners.
Zag Bio also announced the appointment of Jason F. Cole, J.D., as its Chief Executive Officer. Mr. Cole brings over 20 years of experience in the biotechnology industry with a track record of success at public and private biotechnology companies, advancing first-in-class therapies from discovery through approval, executing financings and collaborations and driving operational execution to bring new medicines to patients. His most recent roles were as Chief Executive Officer at SalioGen Therapeutics and as Chief Strategy & Financial Officer and Chief Business Officer of bluebird bio. Previously, Mr. Cole served on the executive leadership teams of Zalicus and CombinatoRx. He holds a J.D. from Columbia University and an A.B. in Government from Dartmouth College.
“It is an exciting opportunity to join Zag Bio’s outstanding team and supportive board and investors to realize the potential to transform the treatment of many autoimmune diseases by targeting the thymus,” said Jason Cole, Chief Executive Officer of Zag Bio. “Despite its integral role in central immune tolerance, the thymus has been inaccessible to drugs of all types. With Zag Bio’s novel approach, we aim to bring a new class of thymus-targeted medicines to help patients and their families, beginning with Type 1 diabetes and expanding to many more autoimmune diseases.”
ZAG-101, a tolerogenic therapy for Type 1 diabetes advancing toward the clinic
Zag Bio is advancing ZAG-101, a bifunctional antibody that delivers pancreatic beta cell antigens to the thymus, as its lead development candidate for Type 1 diabetes. This mechanism is designed to expand antigen-specific thymic Tregs to protect beta cells from destruction and preserve their ability to produce insulin in the pancreas. Zag Bio is initially developing ZAG-101 for patients who are newly diagnosed with Type 1 diabetes or at high risk of developing the disease, with the goal of restoring immune tolerance and preventing autoimmunity. The company has evaluated ZAG-101 in a range of preclinical studies and is initiating IND-enabling studies to advance ZAG-101 into clinical development in late 2026.
World class team of drug developers and biotech company builders, with deep experience in first-in-class therapies and autoimmunity
Zag Bio’s technology is inspired by the work of its co-founder Diane Mathis, PhD, Professor of Immunobiology at Harvard Medical School, whose research lab has made pioneering contributions in the field of immunologic tolerance. The other company co-founders are John Kulman, PhD, a scientific innovator, experienced drug developer, and Chief Scientific Officer of Zag Bio; Jo Viney, PhD, a biotech executive and entrepreneur with deep experience in the field of autoimmune diseases; and Alan Crane, a serial entrepreneur and partner at Polaris Partners who has played a founding and operating role in building numerous successful biotech companies. Collectively, the Zag Bio team brings together biotechnology leaders with decades of industry expertise and track records of advancing innovative therapies for patients.
Zag Bio’s Board of Directors brings together experienced biotechnology entrepreneurs, investors and drug developers:
- Alan Crane, MA, MBA; Chair and Co-Founder, Entrepreneur Partner, Polaris Ventures
- Shelley Chu, MD, PhD; Partner Lightspeed Ventures
- Jason Cole, JD; CEO
- Johannes Fruehauf, MD, PhD; Founding Partner, Mission BioCapital
- Christian Schubert, PhD; Head, AbbVie Ventures
- Sylvia Tobé, PhD; Managing Director, T1D Fund
- Jo Viney, PhD; Co-Founder, CEO, Seismic Therapeutic
About Zag Bio
Zag Bio™ has discovered a novel approach to create thymus-targeted medicines to treat and prevent autoimmune diseases by restoring central immune tolerance. Zag Bio designs bifunctional antibodies that deliver self-antigens to antigen-presenting cells in the thymus to harness the body’s natural process for training immune cells to recognize and tolerate self, halting or preventing autoimmune attacks on the body’s own tissues. Zag Bio’s pipeline includes ZAG-101, its lead program for Type 1 diabetes, as well as discovery programs for thymus-targeted therapies to address other autoimmune diseases and help patients and their caregivers. Located in Cambridge, Massachusetts, Zag Bio’s team includes experienced experts in thymus biology, autoimmune diseases and innovative drug development. Zag Bio is supported by a broad syndicate of investors, including Polaris Partners, Mission BioCapital, AbbVie Ventures, the T1D Fund, Lightspeed Ventures, Sanofi Ventures, KdT Ventures, Regeneron Ventures, Boxer Capital, Pear VC, Codon Capital, Alexandria Venture Investments and Gaingels. For more information, visit www.zagbio.com and follow us on LinkedIn.
Media contact:
Kathryn Morris
The Yates Network LLC
914-204-6412
kathryn@theyatesnetwork.com
- Dr Magovčević-Liebisch is an accomplished pharmaceutical and biotechnology executive with over 25 years of leadership experience
- Dr Ruth McKernan FMedSci CBE, co-founder and Interim CEO, will continue as a Non-executive Director
Cardiff, United Kingdom – 3 November 2025 – Draig Therapeutics (“Draig”), a clinical-stage company aiming to transform the treatment of neuropsychiatric diseases, today announced the appointment of Ivana Magovčević-Liebisch, Ph.D., J.D., as President and Chief Executive Officer (CEO). She succeeds Ruth McKernan, Ph.D., who continues as a Director on the Draig Board of Directors.
Dr Magovčević-Liebisch brings more than 25 years of biopharmaceutical leadership and industry experience. She joins Draig having recently served as President and CEO of Vigil Neuroscience, a company she founded, built and led until its acquisition by Sanofi in August 2025. Under her leadership, Vigil evolved from inception with early preclinical assets through IPO to a clinical-stage organisation within 18 months. Dr Magovčević-Liebisch also raised more than $350 million while advancing Vigil’s lead candidates into clinical development, including the first clinical-stage small molecule TREM2 agonist for the potential treatment of Alzheimer’s disease.
Earlier in her career, Dr Magovčević-Liebisch served as Executive Vice President (EVP) and Chief Business Officer at Ipsen where she was responsible for fuelling the company’s pipeline through executing key strategic transactions. Previously, she was Senior Vice President and Head of Global Business Development for the specialty drug business at Teva Pharmaceutical Industries Ltd. She is a member of the Board of Directors for Acrivon Therapeutics, a publicly traded biotech focused on precision-based cancer therapeutics, and Quanterix Corporation, a publicly traded life sciences company focused on ultra-sensitive biomarker-based diagnostics.
Douglas E. Williams, Chairman of the Board, said: “We are delighted to welcome Ivana as Draig’s new President and CEO. She is a fantastic addition to the team given her strong track record in growing companies and advancing innovative product candidates for patients in need, particularly in the neuroscience space. The Board is confident that these qualities will benefit Draig as it advances the clinical development of DT-101, a differentiated AMPAR positive allosteric modulator, for people with major depressive disorder and its broader pipeline in other neuropsychiatric disorders. On behalf of the board, I would like to thank Ruth for her visionary leadership and the rapid progress made to date and I look forward to her continued service on the Board.”
“I am thrilled to be joining Draig as this exciting stage of its development,” said Ivana Magovčević-Liebisch, President and CEO at Draig Therapeutics. “In just a short period of time, Draig has established a strong foundation, assembled a team with exceptional talent, and developed a clear path to advance its pipeline of ground-breaking therapies for people with neuropsychiatric disorders. This company and its approach to neuropsychiatric disorders has enormous potential to develop best-in-class AMPAR- and GABAAR-based therapeutics, and I cannot wait to start working with the board, leadership and team to bring medicines to people that make a significant positive impact on their lives.”
Ruth McKernan, co-founder, Director of Draig Therapeutics and Interim CEO, said, “I am extremely proud of the work we have done building Draig, raising $140M at Series A and starting our first Phase 2 trial with DT-101. I enthusiastically welcome Ivana, whose exemplary skills will develop the company to deliver transformative medicines for people with neuropsychiatric disorders.”
Dr Magovčević-Liebisch holds a Ph.D. in Genetics from Harvard University and received her J.D. in High Technology Law from Suffolk University Law School. She graduated from Wheaton College with a B.A. in Biology and Chemistry. Dr Magovčević-Liebisch is also active in the non-profit world, serving as a trustee at the Museum of Science (Boston) and the Boston Ballet, and as an overseer at Beth Israel Deaconess Medical Center.
A picture of Dr Magovčević-Liebisch is available on request to draigtx@medistrava.com.
ENDS
For further information:
Mark Swallow, Sandi Greenwood
Draig Therapeutics
About Draig Therapeutics
Draig Therapeutics is a clinical-stage company with a mission to transform treatments in Neuropsychiatry. The company is leveraging its founders’ unique scientific expertise in modulating the core glutamate / GABA pathways that play a critical role in neuropsychiatric diseases to advance a pipeline of groundbreaking therapies designed to address large unmet patient needs, including in Major Depressive Disorder (MDD).
Draig is the Welsh word for ‘dragon’ and it reflects the company’s origins in Wales. The name and logo were inspired by this heritage, reflecting its scientific roots stemming from Cardiff University.
Draig was co-founded by Cardiff University and SV Health Investors, which led the seed financing with ICG, and is backed by other leading healthcare venture firms including Access Biotechnology, Canaan Partners, SR One, Sanofi Ventures and Schroders Capital. For more information, please visit www.draigtherapeutics.com

- SUDO-550 demonstrated favorable safety, tolerability, and once-daily pharmacokinetic profile with full central nervous system (CNS) penetration
- Achieved exposure levels associated with >90% inhibition of TYK2-mediated signaling in both the CNS and peripheral circulation
- Phase 2 study in multiple sclerosis (MS) expected to begin in the first half of 2026
CARMEL, Indiana & LONDON--(BUSINESS WIRE)--Sudo Biosciences, (“Sudo”), a biopharmaceutical company committed to designing and developing best-in-class precision TYK2 (tyrosine kinase 2) inhibitors, today announced positive topline results from its Phase 1 first-in-human clinical trial of SUDO-550, an oral, CNS-penetrant, allosteric TYK2 inhibitor for the treatment of neuroinflammatory diseases. The study evaluated single and multiple ascending doses of SUDO-550 in healthy volunteers, including participants aged 65 and older.
The Phase 1 results demonstrated that SUDO-550 was safe, well tolerated, and suitable for once-daily dosing across a wide dose range. Pharmacokinetic analyses showed dose-proportional exposure, a long half-life, and equivalent free concentrations in plasma and cerebrospinal fluid (CSF), supporting convenient once-daily dosing. Notably, SUDO-550 achieved concentrations over 24-hours consistent with near complete (>90%) inhibition of TYK2-mediated pathways in the peripheral circulation and CNS, including activity in both microglia and astrocytes. Pharmacodynamic assessments confirmed target engagement in both blood and CSF. Results in participants aged 65 and older were consistent with younger participants, supporting suitability for a broad patient population.
“We believe SUDO-550 represents a new generation of selective TYK2 inhibitors engineered to safely reach the brain and modulate the inflammatory processes driving some of the most challenging autoimmune and neurologic diseases,” said Scott Byrd, Chief Executive Officer, Sudo Biosciences. “These results support advancing SUDO-550 into Phase 2 and brings us a step closer to changing the treatment landscape for patients who currently have limited options.”
“The ability of SUDO-550 to access the CNS and achieve concentrations providing sustained inhibition of TYK2 pathways is highly encouraging,” said Dr. Ian Mills, Chief Medical Officer, Sudo Biosciences. “SUDO-550 demonstrated low peak-to-trough variability following once daily dosing, reducing the risk of adverse effects that can be associated with peak drug concentrations, while providing continuous pharmacological activity over 24 hours. This unique clinical profile differentiates SUDO-550 from other TYK2 inhibitors and highlights its potential to advance the standard of care for patients with MS and other neuroinflammatory diseases.”
Sudo Biosciences plans to initiate a Phase 2 study of SUDO-550 in MS in the first half of 2026.
The company is advancing a pipeline of highly selective, small molecule TYK2 pseudokinase inhibitors designed to target the immune and inflammatory pathways that drive autoimmune and neurologic diseases.
About SUDO-550
SUDO-550 is a potential first-in-class and best-in-class, oral, CNS-penetrant allosteric TYK2 inhibitor designed to achieve potent and selective inhibition of TYK2-mediated signaling across both the CNS and peripheral immune systems. In a Phase I clinical study, SUDO-550 was safe and well tolerated and demonstrated a once-daily pharmacokinetic profile with sustained and equivalent exposure levels in the peripheral circulation and CNS. By precisely modulating pathways driven by interleukin-12 (IL-12), interleukin-23 (IL-23), and Type I interferons, SUDO-550 has the potential to treat a broad range of autoimmune and neuroinflammatory diseases, including MS, ALS, and Alzheimer’s disease.
About Sudo Biosciences
Sudo Biosciences is a clinical-stage biopharmaceutical company developing next-generation TYK2 inhibitors to transform the treatment of immune-mediated diseases. Its pipeline includes a potential first- and best-in-class, once-daily oral, brain-penetrant candidate for MS and other neuroinflammatory conditions, as well as a topical candidate for dermatologic diseases. Based in Carmel, IN and London, U.K., Sudo is advancing novel medicines designed to improve patient lives. For more information, visit www.sudobio.com.
Contacts
Media
Kimberly Ha
KKH Advisors
917-291-5744
Kimberly.ha@kkhadvisors.com
Boston, MA, October 28, 2025 - NodThera, a clinical-stage biotech pioneering a paradigm shift in the treatment of chronic inflammatory diseases through NLRP3 inhibition, today announced the appointment of Chris Guiffre, J.D., M.B.A., as the Company’s Chief Financial Officer (CFO).
“We are pleased to welcome Chris to the NodThera team,” said Don Nicholson, Ph.D., Executive Chair of the Board of NodThera. “His successful track record guiding biotechnology companies through significant organizational growth will be an invaluable addition to NodThera as we continue to advance our wholly-owned pipeline of novel NLRP3 inhibitors. On behalf of the entire board and leadership team, I look forward to Chris’ many contributions as we work together to bring our important therapies to patients.”
Chris is a veteran biotech leader with more than 25 years of executive experience, serving in senior leadership roles across public and private life science and tech companies. Before joining NodThera, he served as interim CFO of Vizgen. Earlier, Chris served as President & Chief Operating Officer (COO) of Auron Therapeutics, where he also held CFO responsibilities. Prior, Chris was CFO & COO of Pear Therapeutics and, before that, held Chief Business Officer (CBO), CFO, COO, and Chief Executive Officer (CEO) roles at Cerulean Pharma. During his tenure at Pear and Cerulean, Chris was instrumental in taking both companies public. He also has held senior executive positions at various life science and tech companies, including President & CEO of Alvos Therapeutics, CBO of Hydra Biosciences, Senior Vice President, General Counsel & Secretary at Cubist Pharmaceuticals, and Vice President, General Counsel & Clerk at Renaissance Worldwide. Chris received his J.D. from Boston College Law School, M.B.A. from Boston College Carroll School of Management, and B.S. from Babson College. Chris is additionally an Adjunct Assistant Professor of Management at Bentley University where he teaches a course on strategy.
“I am honored to join NodThera at such an exciting time in the company’s evolution as we advance our broad pipeline with the potential to transform the treatment of cardiometabolic diseases” said Chris Guiffre, CFO of NodThera. “Since its inception, NodThera has built a tremendous reputation for pioneering science, and now the company is pursuing multiple, high-value Phase 2 clinical trials to treat inflammation-driven diseases at the source. I’m eager to work with our entire team to rapidly develop important therapies for patients living with diseases driven by inflammation.”
About NLRP3
NLRP3 activation mediates the release of proinflammatory cytokines, including IL-1β, IL-18 and IL-6, driving chronic inflammation in cardiometabolic, neurological and other diseases. NLRP3 Inhibition offers an alternative approach to therapeutic blockade of individual cytokines like IL-1β, IL-18 and IL-6.
About NodThera
NodThera, a US and UK based biotech developing best-in-class NLRP3 inhibitors, is pioneering a paradigm shift in the treatment of diseases driven by chronic inflammation. NodThera has built an advanced pipeline of multiple clinical-stage small-molecule NLRP3 inhibitors. Compelling first-in-class clinical data, including in patients with cardiometabolic and neurological diseases, is fuelling rapid clinical development towards multiple approvals.
NodThera is backed by top-tier investors including 5AM Ventures, Blue Owl Capital, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners.
NodThera is headquartered in Boston, Massachusetts, with R&D base in the UK. Learn more at www.nodthera.com or follow the Company on LinkedIn.
Investors and Media
Argot Partners
- NT-0796 is an oral, brain-penetrant NLRP3 inhibitor that selectively regulates chronic inflammation, a key driver of cardiometabolic diseases
- Expects to complete the ongoing NT-0796 monotherapy trial (RESOLVE-1) and NT-0796 combination trial with GLP-1 receptor agonist (RESOLVE-2) by 3Q 2026
Boston, MA, October 27, 2025 - NodThera, a clinical-stage biotech pioneering a paradigm shift in the treatment of chronic inflammatory diseases through NLRP3 inhibition, today announced that the first patients have been dosed in its Phase 2 RESOLVE-2 clinical trial (RESolution Of infLammation to treat obesity and cardioVascular disEase) investigating NT-0796, its oral, brain-penetrant NLRP3 inhibitor, in combination with the GLP-1 receptor agonist (GLP-1RA) semaglutide.
“RESOLVE-2 study is a key component of our strategy to develop NT-0796 as a transformative cardiometabolic therapy, as we believe NT-0796 has the potential to reduce cardiovascular risk, to improve metabolic health, and to reduce the burden of related comorbidities,” said Dr. Jyothis George, M.D., Ph.D., FRCP, FACE, Chief Medical Officer of NodThera. “Through our clinical plans, including multiple Phase 2 studies evaluating NT-0796 across cardiometabolic diseases, which will be the longest trials to date for any NLRP3 inhibitor, we seek to reproduce compelling improvements on a range of parameters like IL-6, fibrinogen and hsCRP which reduced rapidly by approximately 80% in patients at high cardiovascular risk.”
RESOLVE-2 is a randomized, double-blind, placebo-controlled Phase 2 clinical trial that will evaluate the safety and efficacy of NT-0796 in combination with GLP-1RA. The trial is expected to enroll approximately 60 people living with obesity, all of whom will receive GLP-1RA, with one half randomized to NT-0796 and the other half to placebo (ClinicalTrials.gov NCT07220629). Similar to RESOLVE-1, the ongoing randomized, double-blind, placebo-controlled Phase 2 clinical trial of NT-0796 monotherapy, endpoints in RESOLVE-2 include changes multiple inflammatory and metabolic biomarkers, safety, tolerability and change in body weight. NodThera expects to complete both trials in 3Q 2026.
"Addressing chronic inflammation in the brain and body remains a substantial unmet need for patients with cardiometabolic diseases," said Melanie Davies, Professor of Diabetes, Diabetes Research Centre, University of Leicester and NIHR Leicester Biomedical Research Centre. “NT-0796 has so far demonstrated a unique ability to target the root cause of obesity and related diseases by reducing residual inflammation. I look forward to RESOLVE-2’s readout next year.”
About NT-0796
NT-0796 is an oral, brain-penetrant NLRP3 inhibitor that selectively regulates chronic inflammation, with an excellent tolerability profile. It is being developed to address the spectrum of cardiometabolic disorders including atherosclerotic cardiovascular disease risk reduction and management of body weight. Clinical data from NodThera’s study in people living with obesity and increased cardiovascular risk demonstrated NT-0796’s anti-inflammatory effect, reducing multiple markers of cardiometabolic risk including high sensitivity CRP reductions comparable to injectable biologics blocking IL-1 and IL-6. In addition, NodThera has published clinical data showing rapid reduction in neuroinflammation, concordant with reductions in peripheral inflammation.
For reduction or maintenance of weight, NT-0796’s mechanism aims to resolve hypothalamic inflammation, thereby restoring multiple dysregulated pathways in obesity, and resetting the body’s set point to defend fat mass. In clinical studies to date, NT-0796 has demonstrated a robust safety and tolerability profile, with no gastrointestinal side effects reported. Preclinical data published in the Journal of Pharmacology and Experimental Therapeutics demonstrate how NLRP3 inhibition has the potential to reverse diet-induced obesity and inflammation, matching weight loss driven by the GLP-1RA semaglutide as a monotherapy. Additionally, in preclinical data published in the journal Obesity, NT-0796’s effect on weight loss was enhanced when combined with semaglutide.
NT-0796 is currently being investigated in two Phase 2 trials in patients living with obesity, as a monotherapy in the RESOLVE-1 trial (ClinicalTrials.gov NCT07055516), including patients with and without type 2 diabetes, and in combination with GLP-1 receptor agonists in the RESOLVE-2 trial (Clinicaltrials.gov NCT07220629).
About NLRP3
NLRP3 activation mediates the release of proinflammatory cytokines, including IL-1β, IL-18 and IL-6, driving chronic inflammation in cardiometabolic, neurological and other diseases. NLRP3 Inhibition offers an alternative approach to therapeutic blockade of individual cytokines like IL-1β, IL-18 and IL-6.
About NodThera
NodThera, a US and UK based biotech developing best-in-class NLRP3 inhibitors, is pioneering a paradigm shift in the treatment of diseases driven by chronic inflammation. NodThera has built an advanced pipeline of multiple clinical-stage small-molecule NLRP3 inhibitors. Compelling first-in-class clinical data, including in patients with cardiometabolic and neurological diseases, is fuelling rapid clinical development towards multiple approvals.
NodThera is backed by top-tier investors including 5AM Ventures, Blue Owl Capital, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners.
NodThera is headquartered in Boston, Massachusetts, with R&D base in the UK. Learn more at www.nodthera.com or follow the Company on LinkedIn.
Investors and Media
Argot Partners

October 2, 2025
EMERYVILLE, Calif. and UTRECHT, The Netherlands and PHILADELPHIA, Oct. 02, 2025 (GLOBE NEWSWIRE) -- XOMA Royalty Corporation (“XOMA Royalty”) (NASDAQ: XOMA) and LAVA Therapeutics N.V. (“LAVA”) (Nasdaq: LVTX) today announced the extension of the expiration of the tender offer to purchase all outstanding shares of common shares of LAVA, for (i) a cash amount to be determined in accordance with the Purchase Agreement, plus (ii) a non-transferable contingent value right (“CVR”) per share representing the right to receive 75% of the net proceeds related to LAVA’s two partnered assets and 75% of any net proceeds from any out license or sale of LAVA’s unpartnered programs (the “Offer”).
The Offer, which was previously scheduled to expire one minute after 11:59 p.m. Eastern Time on October 3, 2025, has been extended until one minute after 11:59 p.m. Eastern time on October 17, 2025, unless the Offer is further extended or earlier terminated. The proposed acquisition is expected to close in the fourth quarter of 2025, subject to customary closing conditions.
LAVA shareholders who previously have tendered their shares do not need to re-tender their shares or take any other action in response to the extension of the Offer. LAVA shareholders have signed support agreements to tender their shares in the Offer prior to the expiration date and support the Offer.
The closing of the Offer is subject to certain conditions, including the tender of LAVA common shares representing at least 80% (or, in certain cases, 75%) of LAVA’s issued and outstanding shares, the condition that certain resolutions are adopted by LAVA’s shareholders meeting, a minimum cash balance at closing, and other customary closing conditions. Following a subsequent offering period, LAVA will undergo a corporate reorganization designed to result in XOMA Royalty acquiring 100% of the shares in LAVA’s successor and all then-remaining LAVA shareholders (other than XOMA Royalty) receiving the same cash and non-transferable contingent value right consideration per share as is provided in the tender offer, subject to applicable withholding taxes. LAVA will hold a shareholder’s meeting in connection with the transactions prior to early November 2025.
About XOMA Royalty Corporation
XOMA Royalty is a biotechnology royalty aggregator playing a distinctive role in helping biotech companies achieve their goal of improving human health. XOMA Royalty acquires the potential future economics associated with pre-commercial and commercial therapeutic candidates that have been licensed to pharmaceutical or biotechnology companies. When XOMA Royalty acquires the future economics, the seller receives non-dilutive, non-recourse funding they can use to advance their internal drug candidate(s) or for general corporate purposes. XOMA Royalty has an extensive and growing portfolio of assets (asset defined as the right to receive potential future economics associated with the advancement of an underlying therapeutic candidate). For more information about XOMA Royalty and its portfolio, please visit www.xoma.com or follow XOMA Royalty Corporation on LinkedIn.
About LAVA Therapeutics
LAVA Therapeutics N.V. is a biopharmaceutical company that has developed several clinical-stage bispecific gamma delta T cell engagers using its proprietary Gammabody® platform, including JNJ-89853413, targeting CD33 and hematologic cancers (NCT06618001), partnered with Johnson & Johnson, and PF-08046052, targeting EGFR and solid tumors (NCT05983133), partnered with Pfizer, Inc. For more information on LAVA, please visit www.lavatherapeutics.com.
Gammabody® is a registered trademark of LAVA Therapeutics N.V.
XOMA Royalty Forward-Looking Statements/Explanatory Notes
Certain statements contained in this press release are forward-looking statements, including statements regarding the expected timing and ability to satisfy the conditions required to close the tender offer, the transactions related to the Purchase Agreement, the ability of XOMA Royalty to monetize LAVA’s programs for the benefit of XOMA Royalty and LAVA shareholders, and the ability to achieve any dispositions within the disposition period under the CVR. In some cases, you can identify such forward-looking statements by terminology such as “anticipate,” “approximately,” “look to,” “plan,” “expect,” “may,” “will,” “could” or “should,” the negative of these terms or similar expressions. These forward-looking statements are not a guarantee of XOMA Royalty’s performance, and you should not place undue reliance on such statements. These statements are based on assumptions that may not prove accurate, and actual results could differ materially from those anticipated due to certain risks including the risk that
XOMA Royalty does not achieve the anticipated benefits from LAVA’s two partnered assets or the potential out license or sale of LAVA’s unpartnered programs, the risk that XOMA Royalty is unable to enter into dispositions related to the LAVA programs, and risks that the conditions to the closing the transaction in the Purchase Agreement are not satisfied. Other potential risks to XOMA Royalty meeting these expectations are described in more detail in XOMA Royalty’s most recent filing on Form 10-Q and in other filings with the Securities and Exchange Commission. Any forward-looking statement in this press release represents XOMA Royalty’s beliefs and assumptions only as of the date of this press release and should not be relied upon as representing its views as of any subsequent date. XOMA Royalty disclaims any obligation to update any forward-looking statement, except as required by applicable law.
EXPLANATORY NOTE: Any references to “portfolio” in this press release refer strictly to milestone and/or royalty rights associated with a basket of drug products in development. Any references to “assets” in this press release refer strictly to milestone and/or royalty rights associated with individual drug products in development.
LAVA’s Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as “anticipate”, “believe”, “could”, “will”, “may”, “expect”, “should”, “plan”, “intend”, “estimate”, “potential”, “suggests”, and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on LAVA’s expectations and assumptions as of the date of this press release and are subject to various risks and uncertainties that may cause actual results to differ materially from these forward-looking statements. As a result, a number of important factors could cause actual results to differ materially from those indicated by such forward-looking statements, including: the risk that the Transactions may not be completed in a timely manner, or at all, which may adversely affect LAVA’s business and the price of its ordinary shares; the delay or failure of the Offer Conditions to be satisfied (or waived), including insufficient ordinary shares of LAVA being tendered in the Offer; the possibility that competing offers will be made; significant costs associated with the Transactions; the risk that any shareholder or other litigation in connection with the Transactions may result in significant costs of defense, indemnification and liability; the risk that activities related to the CVR Agreement may not result in any value to LAVA’s shareholders; the possibility that prior to the completion of the Transactions, LAVA’s or XOMA Royalty’s business may experience significant disruptions due to transaction-related uncertainty; the effects of disruption from the transactions of LAVA’s business and the fact that the announcement and pendency of the Transactions may make it more difficult to establish or maintain relationships with employees, manufacturers, suppliers, vendors or business partners; the occurrence of any event, change or other circumstance that could give rise to the termination of the Purchase Agreement; as well as potential adverse effects on LAVA’s business condition and results from general economic and market conditions and overall fluctuations in the United States and international equity markets, including as a result of inflation, heightened interest rates, recent and potential future pandemics and other health crises, and hostilities, including the Russian invasion of Ukraine and the conflict in the Middle East. These and other risks are described in greater detail under the caption “Risk Factors” in LAVA’s most recent Annual Report on Form 10-K and other filings LAVA makes with the U.S. Securities and Exchange Commission (the “SEC”). LAVA assumes no obligation to update any forwardlooking statements contained herein whether as a result of any new information, future events, change in expectations or otherwise, except as otherwise required by law.
Additional Information and Where to Find It
The description contained in this press release is for informational purposes only and is not a recommendation, an offer to buy or the solicitation of an offer to sell any shares of LAVA’s ordinary shares. The tender offer for LAVA’s outstanding ordinary shares described in this report has been extended until one minute after 11:59 p.m. Eastern time on October 17, 2025, unless the Offer is further extended or earlier terminated. XOMA Royalty will file an amendment to its Tender Offer Statement on Schedule TO with the SEC. LAVA filed an updated Solicitation/Recommendation Statement on Schedule 14D-9 with the SEC related to the tender offer. LAVA also filed a proxy statement in connection with the originally scheduled EGM at which LAVA’s shareholders were requested to vote on certain proposed resolutions (the “EGM Proposals”) in connection with the Transactions. LAVA plans to send a revised proxy statement and proxy card to each shareholder entitled to vote at the reconvened extraordinary general meeting.
INVESTORS AND SECURITY HOLDERS ARE URGED TO READ THE PROXY STATEMENT REGARDING THE EXTRAORDINARY GENERAL MEETING AND THE TENDER OFFER MATERIALS (INCLUDING THE OFFER TO PURCHASE, A LETTER OF TRANSMITTAL AND RELATED DOCUMENTS) AND THE SOLICITATION/RECOMMENDATION STATEMENT ON SCHEDULE 14D-9 REGARDING THE OFFER, AS THEY MAY BE AMENDED OR SUPPLEMENTED FROM TIME TO TIME, WHEN THEY BECOME AVAILABLE BECAUSE THEY WILL CONTAIN IMPORTANT INFORMATION THAT INVESTORS AND SECURITY HOLDERS SHOULD CONSIDER BEFORE MAKING ANY DECISION REGARDING TENDERING THEIR SHARES (INCLUDING THE TERMS AND CONDITIONS OF THE OFFER) OR MAKING ANY VOTING DECISION FOR THE EXTRAORDINARY GENERAL MEETING.
Investors and security holders may obtain a free copy of these statements (when available) and other documents filed with the SEC at the website maintained by the SEC at www.sec.gov or by directing such requests to the information agent for the Offer, which is named in the tender offer statement. Investors and security holders may also obtain, at no charge, the documents filed or furnished to the SEC by LAVA under the “SEC Filings” subsection of the “Financials & Filings” section of LAVA’s website at https://ir.lavatherapeutics.com or by accessing the Investor Relations sections of XOMA Royalty’s website at https://www.investors.xoma.com.
Participants in the Solicitation
LAVA, its directors and executive officers, and other members of its management and employees, as well as XOMA Royalty and its directors and executive officers, may be deemed to be participants in the solicitation of proxies from LAVA’s shareholders in connection with the EGM Proposals. Information about LAVA’s directors and executive officers and their ownership of Shares is set forth in the proxy statement for LAVA’s 2025 annual general meeting of shareholders, which was filed with the SEC on April 28, 2025. Information about XOMA Royalty’s directors and executive officers is set forth in the proxy statement for XOMA Royalty’s 2025 annual meeting of shareholders, which was filed with the SEC on April 15, 2025. Shareholders may obtain additional information regarding the direct and indirect interests of the participants in the solicitation of proxies in connection with the EGM Proposals, including the interests of LAVA’s directors and executive officers in the Transactions, which may be different than those of LAVA’s shareholders generally, by reading the proxy statement and other relevant documents regarding the Transactions which will be filed with the SEC.
XOMA Royalty Investor Contact
Juliane Snowden
XOMA Royalty Corporation
+1 646-438-9754
XOMA Royalty Media Contact
Kathy Vincent
KV Consulting & Management
+1 310-403-8951
LAVA Therapeutics Investor Contact
Fred Powell
LAVA Therapeutics
+1 800-311-6892

- Funding to advance EXPD-101, a Phase 2–ready, once-daily, oral DPP1 inhibitor with first-in-class potential into a global development program in COPD, and a broad spectrum of other neutrophil-driven diseases.
- Financing co-led by Sofinnova Investments and Novo Holdings, with participation from Forbion, Dawn Biopharma (a platform controlled by KKR), Adage, Balyasny, Logos Capital, Sanofi Ventures, and existing investors BVF Partners and Venrock Healthcare Capital Partners.
- Andrew Cheng, M.D., Ph.D., CEO of Akero Therapeutics and former CMO of Gilead Sciences, named Chairman of the Board.
SAN FRANCISCO, CA – October 9, 2025 - Expedition Therapeutics, a biotech company developing therapies for novel inflammatory and respiratory diseases, today announced the closing of an oversubscribed and upsized $165 million Series A financing. The round was co-led by Sofinnova Investments and Novo Holdings, with additional investment from Forbion, Dawn Biopharma (a platform controlled by KKR), Adage, Balyasny, Logos Capital, Sanofi Ventures, and existing investors BVF Partners and Venrock Healthcare Capital Partners.
Proceeds will advance Expedition’s lead candidate, EXPD-101, through a global Phase 2 study in chronic obstructive pulmonary disease (COPD). Current COPD therapies offer limited benefits, particularly in non-type 2 COPD, which affects nearly 70% of patients. The funding will also support indication expansion, positioning the program with first-in-class and best-in-class potential across a broad range of neutrophil-driven inflammatory diseases.
EXPD-101 is a next-generation DPP1 inhibitor with first-in-class potential. It is designed to target neutrophilic inflammation, which is a key underlying driver of COPD across the disease spectrum. In Phase 1 studies, EXPD-101 was well tolerated with no dose- limiting toxicities and demonstrated clear target engagement with pharmacokinetics supporting a once-daily oral dosing.
“Most COPD patients lack effective treatment options,” said Yi Larson, Founder and CEO, Expedition Therapeutics. “DPP1 represents an exciting new mechanism for COPD, and EXPD-101 has the potential to be a first-in-class therapy with this target. This financing from top-tier investors allows us to accelerate development, with the potential to treat a broad range of COPD patients, addressing a major unmet need and improving the standard of care for millions. Beyond COPD, EXPD-101 also has first/best-in-class potential to address a broad spectrum of other neutrophil-driven diseases in the future.”
In August 2025, Expedition acquired exclusive worldwide rights, excluding mainland China, Hong Kong and Macau, from Fosun Pharma to develop and commercialize EXPD-101 in all indications. EXPD-101 is currently advancing in a Phase 2 bronchiectasis study led by partner Fosun Pharma within China.
Expedition is led by a management team with deep experience in respiratory therapeutics, drug development, and corporate strategy:
- Yi Larson, Founder and CEO, former CFO of LianBio and Turning Point Therapeutics. Over $100 billion in M&A and financing transactions at Goldman Sachs, and board roles at RayzeBio and Olema Oncology.
- Geoff Gilmartin, M.D., CMO, with leadership experience at AstraZeneca and Vertex and formerly Chief Medical Officer of Proteostatis. Most recently Geoff led the benralizumab (Fasenra) program across multiple indications including COPD and asthma.
- Eric Hu, Ph.D., Chief Business Officer, with 20 years of R&D and business development experience at Overland Pharma, Turning Point Therapeutics, Gilead Sciences, and Mitsubishi Tanabe.
The company is advised by a world-class scientific committee led by James Chalmers, M.D., Ph.D.; Alvar Agustí, M.D., Ph.D.; Mark Dransfield, M.D.; Dave Singh, M.D.; and Surya Bhatt, M.D. This team of globally recognized COPD leaders brings decades of clinical and translational expertise.
“Expedition is led by a proven team with deep expertise in respiratory drug development and a strategy that could deliver a disease controlling therapy for COPD,” said Jonathan Leff, M.D., Executive Partner, Sofinnova Investments. “We are excited to partner with the company to advance this important mission.”
“COPD is one of the largest and fastest growing healthcare challenges globally, and Expedition’s unique approach has the potential to significantly improve patient outcomes,” said Ken Harrison, Ph.D., Senior Partner, Novo Holdings. “We look forward to supporting the company’s growth as it advances EXPD-101 in the clinic.”
Joining the Expedition Board of Directors as part of this financing are Andrew Cheng, M.D., Ph.D. (CEO, Akero Therapeutics; former CMO, Gilead Sciences), Erin Lavelle (former COO/CFO, ProfoundBio), Regina Salvat, Ph.D. (Principal, Forbion), Jonathan Leff, M.D. (Executive Partner, Sofinnova Investments), Ken Harrison, Ph.D. (Senior Partner, Novo Holdings), and Gorjan Hrustanovic, Ph.D. (Partner, BVF Partners L.P.) alongside Yi Larson, Founder & CEO, Expedition Therapeutics.
About Expedition Therapeutics
Expedition Therapeutics is a biotechnology company developing novel therapies for serious inflammatory and respiratory diseases. The company’s lead program, EXPD-101, is a next-generation once-daily DPP1 inhibitor with first-in-class potential in COPD and first/best-in-class potential across a broad spectrum of other neutrophil-driven diseases. Expedition combines deep translational expertise, clinical development experience, and a platform for in-licensing global innovation. For more information, visit www.expeditiontx.com.
Media Contact
Kimberly Ha
KKH Advisors
kimberly.ha@kkhadvisors.com
917-291-5744
- Sanofi and QuantHealth team up to advance AI-powered digital twins and clinical trial
- simulations to accelerate drug development
Tel Aviv & Cambridge, MA – October 1st, 2025 — QuantHealth, a pioneer in AI-driven clinical trial simulation, announced a strategic investment from Sanofi Ventures, the venture capital arm of global healthcare leader Sanofi. The investment will accelerate QuantHealth’s efforts to bring scalable, patient-level simulations and digital twin technologies to the forefront of drug development, bringing its total funds raised to $30 million.
QuantHealth’s platform enables pharmaceutical companies to virtually simulate clinical trials using real-world data and advanced AI models. By generating millions of patient-level digital twins, the platform predicts trial outcomes and optimizes protocol design, aiming to improve trial success rates, timelines, and cost efficiency.
The investment has further catalyzed an enterprise strategic relationship that supports Sanofi’s focus on integrating advanced AI and digital technologies to drive transformation across its R&D ecosystem. As part of the investment, Cris De Luca, Partner at Sanofi Ventures, will join as an observer on QuantHealth’s Board of Directors.
“We’re proud to welcome Sanofi Ventures as an investor and strategic ally in our journey,” said Orr Inbar, CEO and Co-founder of QuantHealth. “Sanofi is at the forefront of digital transformation in pharma, and this relationship will help us scale our impact and bring more predictive, AI-driven approaches to clinical development.”
“QuantHealth has the potential to transform how clinical trials are designed and optimized,” said Cris De Luca, Partner at Sanofi Ventures. “Their approach to leveraging digital twins and real-world data is advancing the next generation of R&D, and I look forward to supporting the team.”
“At Sanofi, we are building an AI-first organization, and engaging with innovators like QuantHealth is central to our strategy,” said Emmanuel Frenehard, Chief Digital Officer at Sanofi. “Their platform represents an important opportunity to reimagine clinical trial design through simulation, and we’re excited to explore this capability together.”
This investment and enterprise relationship underscores a shared commitment to advancing drug development through the responsible use of artificial intelligence in life sciences.
About QuantHealth
QuantHealth is an AI-driven clinical trial simulation company that helps pharmaceutical companies
design faster, more successful trials using real-world data and predictive modeling. With access to over 350 million patient records and proprietary AI algorithms, QuantHealth’s platform simulates trials at scale to optimize protocols, reduce risk, and accelerate timelines.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi, investing globally in early-stage biotech and digital health companies aligned with Sanofi’s mission to chase the miracles of science. Areas of focus include immunology, oncology, rare diseases, vaccines, and digital innovation.
Proceeds to support continued clinical advancement of lead program, VGA039, a first-in-class antibody targeting protein S for the treatment of bleeding disorders, starting with von Willebrand disease (VWD)
VGA039 is the first subcutaneous therapy, dosed once monthly, that can address all types of VWD and all types of bleeds; pivotal Phase 3 trial of VGA039 in VWD patients initiated
Financing co-led by Sanofi Ventures and Viking Global Investors, with participation from other new investors including Janus Henderson Investors, Frazier Life Sciences and GordonMD Global Investments
SOUTH SAN FRANCISCO, CA, September 30, 2025 – Star Therapeutics, a clinical stage biotechnology company discovering and developing best-in-class antibodies for bleeding disorders and other diseases, today announced an oversubscribed $125 million Series D financing. The financing round was co-led by Sanofi Ventures and Viking Global Investors with participation from both existing and other new investors, including Janus Henderson Investors, Frazier Life Sciences and GordonMD Global Investments. Existing investors include Agent Capital, Blue Owl Capital, Catalio Capital Management, Cormorant Asset Management, New Leaf Venture Partners, NYBC Ventures, OrbiMed, Qatar Investment Authority (QIA), RA Capital Management, Redmile Group, Sofinnova Investments, Soleus Capital and Westlake BioPartners.
Proceeds from the Series D financing will support the continued advancement of Star’s pipeline including its lead program, VGA039, a first-in-class monoclonal antibody that targets Protein S to restore balance in blood clotting. VGA039 is being developed as a universal hemostatic therapy for multiple bleeding disorders, starting with von Willebrand disease (VWD). Star has initiated a pivotal Phase 3 clinical trial of VGA039 in patients with all types of VWD, designed to evaluate VGA039 administered once monthly via subcutaneous injection.
“We’re making significant progress across our pipeline, highlighted by the recent initiation of our Phase 3 trial of VGA039 to prevent bleeding in people with all types of VWD, the most common inherited bleeding disorder, with more than 50,000 patients diagnosed and treated in the U.S.,” said Adam Rosenthal, Ph.D., CEO and Founder of Star Therapeutics. “VGA039 has the potential to be transformative for VWD patients and could meaningfully reduce the treatment burden, given its once monthly subcutaneous dosing regimen, in comparison to factor replacement prophylaxis which requires two to three IV infusions per week. This new funding from leading life sciences investors reinforces the urgency and impact of our mission: to bring life-changing therapies to people living with serious bleeding disorders and other diseases in hematology and immunology.”
In conjunction with the financing, Jason Hafler, Ph.D., from Sanofi Ventures will join Star’s board of directors, and Maneka Mirchandaney of Viking Global Investors will join as a board observer.
“At Sanofi Ventures, we’re proud to bring meaningful experience in supporting innovative therapies for bleeding disorders and remain deeply committed to exploring best-in-class therapies to better meet the needs of patients. VWD represents a compelling opportunity, with a sizable patient population and significant unmet need,” said Jason Hafler, Ph.D., Managing Director at Sanofi Ventures. “With VGA039 advancing into Phase 3 for VWD, Star is wellpositioned to deliver both substantial patient impact and long-term value. We’re proud to support a team with a strong track record and a bold vision for transforming care in VWD and beyond.”
About VGA039
VGA039 is a monoclonal antibody therapy with a novel mechanism of action that targets Protein S, thereby restoring balance to the blood clotting process. VGA039 has potential to be a universal hemostatic therapy that can treat numerous bleeding disorders, starting with VWD. As a subcutaneously self-administered antibody therapy with a convenient once monthly dosing regimen, VGA039 has the potential to dramatically reduce treatment burden for patients. VGA039 has received Fast Track and orphan drug designations from the United States Food and Drug Administration (FDA).
Interim positive data from VIVD-2, a Phase 1 single ascending dose study of VGA039 in patients with VWD, were previously reported at the Annual Meeting of the American Society of Hematology (ASH) in December 2024. A Phase 1/2 multidose study, VIVID-3, is ongoing (NCT05776069). VGA039 has advanced into a Phase 3 study, VIVID-6 (NCT07115004), a global single arm cross-over study designed to investigate the safety and efficacy of subcutaneous administration of VGA039 as prophylaxis for bleeding in patients with every type of VWD. For additional information on VIVID-6, including how to enroll, please visit the website here.
About von Willebrand disease
Von Willebrand disease (VWD) is the most common inherited bleeding disorder in which the blood does not clot properly, caused by absent or defective von Willebrand factor (VWF). VWD patients may experience excessive bleeding with variability in severity and frequency, negatively impacting their daily lives. Current therapies for VWD prophylaxis include factor replacement therapies requiring multiple intravenous (IV) infusions every week. More than 50,000 people in the United States are estimated to be receiving treatment for VWD.
About Star Therapeutics
Star Therapeutics is a clinical stage biotechnology company discovering and developing best-inclass antibodies to create life-changing therapies for patients, initially addressing unmet needs in hematology and immunology. The company applies its expertise in antibody innovation to interrogate areas of biology that have been overlooked and have the potential to address multiple diseases with a single therapy. Star’s leadership team has deep expertise in novel antibody drug development, having invented four first-in-class antibodies including the first approved drug (ENJAYMO®) for cold agglutinin disease, a hematology disease, and other therapies that have demonstrated clinical proof of concept. Based in South San Francisco, Star has raised more than $300 million from leading life sciences investors. For more information, please visit StarTherapeutics.com and follow us on LinkedIn and X.
Contacts:
Media:
1AB
Katie Engleman
katie@1abmedia.com
Investors:
Star Therapeutics
Scott Robertson
scott@star-therapeutics.com

- The fund will remain focused on Sanofi's key areas of immunology, rare diseases, neurology, and vaccines, backing earlier-stage innovation and emerging opportunities that support the company’s long-term strategy
- Sanofi Ventures drives innovation through leading and participating in private financing rounds for pioneering healthcare companies
Paris, September 24, 2025. Sanofi Ventures has announced an additional $625 million multiyear capital commitment from Sanofi, increasing its total assets under management to over $1.4 billion. This new commitment to the evergreen venture fund builds on more than a decade of investing in innovative biotech and digital health companies that align with Sanofi’s long-term growth ambitions.
“This new, significant capital commitment reflects our strong belief that some of the most important medical breakthroughs begin in early-stage companies. With a proven track record of strategic wins and successful exits, Sanofi Ventures has become a powerful engine for scientific progress and strategic growth,” said Paul Hudson, Chief Executive Officer at Sanofi. “By strengthening our investment capabilities, we are accelerating our ability to bring next-generation therapies that improve people’s lives while building valuable partnerships across the healthcare ecosystem.”
Sanofi Ventures is the corporate venture capital arm of Sanofi, investing in top-tier biotech and artificial intelligence/digital health companies that focus on helping patients and transforming the practice of medicine. Since its inception in 2012, the fund has deployed over $800 million across more than 70 innovative companies in biotech and digital health. The team leads investments across all stages of the private company lifecycle, from seed to crossover, serves on boards, and participates in IPOs. Sanofi Ventures maintains a diverse, globally distributed portfolio of companies advancing innovation around the world.
"The increased capital commitment enables us to deepen our role as a leading strategic investor in breakthrough science and digital innovation. Our global portfolio spans the full company lifecycle from seed-stage through IPO participation, demonstrating the long-term value we bring to both entrepreneurs and Sanofi,” said Jason P. Hafler, PhD, Managing Director at Sanofi Ventures. “The strong performance of our fund, including three realized exits in 2024 from companies with a combined acquisition value of $3.25 billion, validates our evergreen structure and approach to identifying and supporting companies that are at the forefront of medical innovation."
Sanofi Ventures' expanded capital commitment comes at a time when access to early-stage funding has become more limited across the biotech sector, positioning the fund to play a crucial role in advancing healthcare innovation. By providing essential financial backing and strategic support, the fund is helping to bridge the gap for promising startups, enabling them to advance potentially life-changing therapies through key development milestones. This strategy not only strengthens Sanofi's pipeline of future innovations but also reinforces the company's position as a leading catalyst for breakthrough science in healthcare.
About Sanofi
Sanofi is an R&D driven, AI-powered biopharma company committed to improving people's lives and delivering compelling growth. We apply our deep understanding of the immune system to invent medicines and vaccines that treat and protect millions of people around the world, with an innovative pipeline that could benefit millions more. Our team is guided by one purpose: we chase the miracles of science to improve people's lives; this inspires us to drive progress and deliver positive impact for our people and the communities we serve, by addressing the most urgent healthcare, environmental, and societal challenges of our time.
Sanofi is listed on EURONEXT: SAN and NASDAQ: SNY
Media Relations
Sandrine Guendoul | +33 6 25 09 14 25 | sandrine.guendoul@sanofi.com
Evan Berland | +1 215 432 0234 | evan.berland@sanofi.com
Léo Le Bourhis | +33 6 75 06 43 81 | leo.lebourhis@sanofi.com
Victor Rouault | +33 6 70 93 71 40 | victor.rouault@sanofi.com
Timothy Gilbert | +1 516 521 2929 | timothy.gilbert@sanofi.com
Léa Ubaldi | +33 6 30 19 66 46 | lea.ubaldi@sanofi.com
Investor Relations
Thomas Kudsk Larsen | +44 7545 513 693 | thomas.larsen@sanofi.com
Alizé Kaisserian | +33 6 47 04 12 11 | alize.kaisserian@sanofi.com
Felix Lauscher | +1 908 612 7239 | felix.lauscher@sanofi.com
Keita Browne | +1 781 249 1766 | keita.browne@sanofi.com
Nathalie Pham | +33 7 85 93 30 17 | nathalie.pham@sanofi.com
Tarik Elgoutni | +1 617 710 3587 | tarik.elgoutni@sanofi.com
Thibaud Châtelet | +33 6 80 80 89 90 | thibaud.chatelet@sanofi.com
Yun Li | +33 6 84 00 90 72 | yun.li3@sanofi.com
Sanofi forward looking statement
This press release contains forward-looking statements as defined in the Private Securities Litigation Reform Act of 1995, as amended. Forward-looking statements are statements that are not historical facts. These statements include projections and estimates and their underlying assumptions, statements regarding plans, objectives, intentions, and expectations with respect to future financial results, events, operations, services, product development and potential, and statements regarding future performance. Forward-looking statements are generally identified by the words “expects”, “anticipates”, “believes”, “intends”, “estimates”, “plans” and similar expressions. Although Sanofi’s management believes that the expectations reflected in such forward-looking statements are reasonable, investors are cautioned that forward-looking information and statements are subject to various risks and uncertainties, many of which are difficult to predict and generally beyond the control of Sanofi, that could cause actual results and developments to differ materially from those expressed in, or implied or projected by, the forward-looking information and statements. These risks and uncertainties include among other things, the uncertainties inherent in research and development, future clinical data and analysis, including post marketing, decisions by regulatory authorities, such as the FDA or the EMA, regarding whether and when to approve any drug, device or biological application that may be filed for any such product candidates as well as their decisions regarding labelling and other matters that could affect the availability or commercial potential of such product candidates, the fact that product candidates if approved may not be commercially successful, the future approval and commercial success of therapeutic alternatives, Sanofi’s ability to benefit from external growth opportunities, to complete related transactions and/or obtain regulatory clearances, risks associated with intellectual property and any related pending or future litigation and the ultimate outcome of such litigation, trends in exchange rates and prevailing interest rates, volatile economic and market conditions, cost containment initiatives and subsequent changes thereto, and the impact that global crises may have on us, our customers, suppliers, vendors, and other business partners, and the financial condition of any one of them, as well as on our employees and on the global economy as a whole. The risks and uncertainties also include the uncertainties discussed or identified in the public filings with the SEC and the AMF made by Sanofi, including those listed under “Risk Factors” and “Cautionary Statement Regarding Forward-Looking Statements” in Sanofi’s annual report on Form 20-F for the year ended December 31, 2024. Other than as required by applicable law, Sanofi does not undertake any obligation to update or revise any forward-looking information or statements.
All trademarks mentioned in this press release are the property of the Sanofi.

JERSEY CITY, NJ – September 17, 2025 – Character Biosciences, a precision medicine company transforming drug development for polygenic diseases, today announced the expansion of its leadership team with four senior appointments, including Robert Kim, MD, MBA as Chief Medical Officer, Daniel Elgort, PhD as Chief Data & Analytics Officer, Josh Buddle as Head of Clinical Network and Longitudinal Research, and Jessamyn Wead as Head of People.
The company also announced an expansion of its Series B financing, bringing total capital raised in the round to more than $110 million. The additional close included participation from Sanofi Ventures, as well as new and existing investors.
“These appointments and the additional funding mark an important moment for Character Bio as we advance our first therapeutic programs toward the clinic,”said Cheng Zhang, Chief Executive Officer of Character Biosciences. “The expanded leadership team and strong support from our investors strengthen our ability to deliver on our mission of transforming outcomes for patients with vision-threatening diseases.”
Proceeds from the financing will enable Character Bio to further advance the clinical development of CTX114, a complement inhibitor for geographic atrophy (GA), and CTX203, a lipid modulator designed to prevent progression to advanced age-related macular degeneration (AMD). This financing follows Character Bio’s previously announced $93 million Series B, led by aMoon Growth Fund and the Luma Group, alongside participation from Bausch + Lomb, Innovation Endeavors, Catalio Capital Management, S32, and additional leading life sciences investors.
“I am thrilled to join Character at such a pivotal time,”said Robert Kim, MD, MBA, Chief Medical Officer of Character Biosciences. “Dry AMD is the next frontier in ophthalmology, and Character’s multidisciplinary approach has the potential to deliver treatments that truly address the unmet needs of patients.”
Jason Hafler, Managing Director of Sanofi Ventures, who will join Character Bio’s Board of Directors, added: “Character has built a compelling portfolio of precision medicine programs and a unique patient data platform. We are excited to partner with the team and contribute to the continued development of their pipeline.”
The new executives join existing leaders Cheng Zhang, CEO, Laura Carter, PhD, CSO, Erik Karrer, PhD, SVP, Drug Discovery, and Kaila Smilen, MBA, Senior Director, Business Development & Corporate Strategy.
About Character Biosciences
Character Biosciences is a precision medicine company developing innovative therapies for progressive polygenic diseases, starting with ophthalmology. By integrating genomics, deep clinical data, and AI-driven modeling, Character Bio identifies disease drivers for targeted patient subtypes to improve the success rate of drug development. The company is advancing a pipeline focused on dry age-related macular degeneration (AMD)and primary open-angle glaucoma (POAG)to address significant unmet medical needs. For more information, visit www.characterbio.com.
Media Contact (Character Biosciences)
Monique Allaire
THRUST Strategic Communications
monique@thrustsc.com
BOSTON – September 10th, 2025 — Odyssey Therapeutics, Inc., a clinical-stage biopharmaceutical company seeking to transform the standard of care for patients suffering from autoimmune and inflammatory diseases by developing medicines that precisely target disease pathology, today announced the closing of a $213 million Series D financing. In addition to participation from all existing investors, new investors included Affinity Asset Advisors, Dimension Capital, Jeito Capital, Lightspeed Ventures, TPG Life Sciences Innovations, and Wedbush Healthcare Partners. Proceeds will support the advancement of Odyssey’s portfolio of clinical and preclinical medicines designed to precisely treat the drivers of complex autoimmune diseases.
“As we advance our programs towards important clinical milestones, we’re focused on translating our scientific efforts into meaningful benefit for patients,” said Gary D. Glick, Ph.D., Founder and CEO of Odyssey Therapeutics. “I’m thankful for the continued support from our investors and their enthusiasm for our mission of delivering truly transformative medicines to patients suffering from inflammatory diseases.”
In addition to the fundraise, Shelley Chu, M.D., Ph.D., Partner at Lightspeed Ventures, Paulina Hill, Ph.D., Partner at Sanofi Ventures, Nan Li, Founder and Managing Partner at Dimension Capital, Carolyn Ng, Ph.D., Business Unit Partner with TPG Life Sciences Innovations, and Ksenija Pavletic, Partner and CCO at Jeito Capital, will join Odyssey’s Board of Directors.
“This financing is a testament to the important medicines Odyssey is working to discover and deliver to patients,” said Jeff Leiden, M.D., Ph.D., Chairman of the Odyssey Board. “I am delighted to welcome our new investors and work alongside our new board members as Odyssey continues to advance its programs through clinical development.”
Since its founding in 2021, Odyssey has built comprehensive drug discovery and development capabilities. The company has driven an internally discovered program from ideation through multiple clinical milestones, nominated multiple development candidates, and established several collaborations with pharmaceutical leaders and biotech innovators.
About Odyssey Therapeutics
Odyssey Therapeutics is a clinical-stage biopharmaceutical company seeking to transform the standard of care for patients suffering from autoimmune and inflammatory diseases by developing medicines that are designed to precisely target disease pathology. Since its founding in 2021, Odyssey has built a portfolio of completely internally discovered and developed medicines with the first reaching multiple clinical milestones in just over two years. The portfolio leverages the scientific expertise of its team of experienced drug hunters and a comprehensive suite of tools to efficiently advance product candidates that it believes have the potential to induce deep and durable remission for patients across several inflammatory diseases with unmet need. For more information, please visit odysseytx.com and follow Odyssey Therapeutics on X and LinkedIn.
About Dimension Capital
Dimension is a multi-stage investment firm dedicated to the interface of technology and the life sciences. The firm partners with visionary founders to build enduring, category- defining businesses across software, infrastructure, therapeutics, diagnostics, and clinical development. Dimension invests in companies around the globe and has offices in New York and San Francisco. Learn more at www.dimensioncap.com.
About Jeito Capital
Jeito Capital is a global leading Private Equity fund with a patient benefit driven approach that finances and accelerates the development and growth of ground-breaking medical innovation. Jeito empowers and supports managers through its expert, integrated, multi- talented team and through the investment of significant capital to ensure the growth of companies, building market leaders in their respective therapeutic areas with accelerated patients' access globally, especially in Europe and the United States. Jeito has built a diversified portfolio of clinical biopharmas with cutting-edge innovations addressing high unmet needs. Jeito Capital is based in Paris with a presence in Europe and the United States. For more information visit: www.jeito.life or follow us on LinkedIn.
About Lightspeed Ventures
Lightspeed Venture Partners is a multi-stage venture capital firm focused on accelerating disruptive innovations and trends in the Enterprise, Consumer, Healthcare, and Fintech sectors. Over the past two decades, the Lightspeed team has backed hundreds of entrepreneurs and helped build more than 500 companies globally including Affirm, Carta, Cato Networks, Epic Games, Faire, Forty Seven, Guardant Health, Mulesoft, Navan, Netskope, Nutanix, Rubrik, Scorpion Therapeutics, Sharechat, Snap, Udaan, Xaira Therapeutics, and more. Lightspeed and its global team currently manage $25B in AUM across the Lightspeed platform, with investment professionals and advisors in the U.S., Europe, India, Israel, and Southeast Asia. https://lsvp.com.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi, focused on investing in promising early-stage healthcare companies. The firm supports pioneering innovations in biotechnology, digital health, and life sciences aligning with Sanofi’s mission to bring life- changing treatments to patients worldwide. For more information visit: www.sanofiventures.com.
About TPG
TPG is a leading global alternative asset management firm, founded in San Francisco in 1992, with $261 billion of assets under management and investment and operational teams around the world. TPG invests across a broadly diversified set of strategies, including private equity, impact, credit, real estate, and market solutions, and our unique strategy is driven by collaboration, innovation, and inclusion. Our teams combine deep product and sector experience with broad capabilities and expertise to develop differentiated insights and add value for our fund investors, portfolio companies, management teams, and communities. For more information, visit www.tpg.com.
- Collaboration leverages OMass’ OdyssionTM platform for the continued development of oral small molecules against a first-in-class target in inflammatory bowel disease
- OMass to receive $20 million upfront payment, with potential for more than $400 million in additional milestone payments, as well as tiered royalties on net sales
Oxford, United Kingdom – 2nd September 2025 2025 – OMass Therapeutics (‘OMass’ or ‘the Company’), a biotechnology company identifying medicines against highly validated target ecosystems such as membrane proteins or intracellular complexes, today announces that it has entered into an exclusive collaboration and license agreement with Genentech, a member of the Roche Group, for the rights to develop and commercialize OMass’ preclinical oral small molecule program for inflammatory bowel disease.
“Using our OdyssIONTM platform, we’ve been able to make significant progress on this novel first-in-class target with a differentiated mechanism of action in inflammatory bowel disease,” said Ros Deegan, CEO of OMass Therapeutics. “Genentech brings a strong legacy of innovation in immunology and world class scientific expertise, making them an ideal partner for this program. We are delighted to be partnering with them and build on the progress we have made to date.”
"There are nearly eight million people living with IBD who are in need of innovative treatment approaches," said Boris L. Zaïtra, Head of Roche Corporate Business Development. "Despite recent advancements, there is still a high unmet medical need which fuels our commitment to partnering with companies such as OMass Therapeutics focused on innovation to accelerate potentially transformative medicines and advance science."
Under the terms of the agreement, OMass will receive an upfront payment of $20 million, plus additional potential preclinical, development, commercial and net sales milestone payments of more than $400 million. OMass is also eligible for tiered royalties on net sales. Under the collaboration, OMass will lead the initial preclinical development of the program until candidate selection. Genentech will be responsible for clinical development, regulatory activities, manufacturing and commercialization.
-ENDS-
For further information, please contact:
|
OMass Therapeutics |
|
|
Rosamond Deegan, Chief Executive Officer Email: ros.deegan@omass.com |
|
|
ICR Healthcare (For media) |
|
|
Namrata Taak / Ashley Tapp Email: omass@icrhealthcare.com |
|
|
Thrust Strategic Communications (For IR) Renee Leck Email: renee@thrustsc.com |
|
About OMass Therapeutics
OMass Therapeutics is a biotechnology company discovering medicines against highly-validated target ecosystems, such as membrane proteins or intracellular complexes.
OdyssION™, OMass’ unique drug discovery platform, comprises next-generation native mass spectrometry with novel biochemistry techniques and custom chemistry to interrogate not just a drug target, but also the interaction of the target with its native ecosystem, separate from the confounding complexity of the cell. This unique approach results in cell-system fidelity with cell-free precision.
OMass is advancing a pipeline of small molecule therapeutics in rare diseases and immunological conditions. Its lead program is a best-in-class MC2 (melanocortin-2) receptor antagonist for the treatment of Congenital Adrenal Hyperplasia (CAH) and ACTH-dependent Cushing syndrome. The focus of the program has been to increase receptor residency time to make OMass’ antagonists resistant to competition by the endogenous ligand, thereby avoiding loss of efficacy in the face of rising adrenocorticotropic hormone (ACTH) levels due to reductions in glucocorticoid supplementation for CAH or progression of Cushing’s Syndrome.
Headquartered in Oxford, UK, OMass has raised over $160M (£129M) from a top-tier international investor syndicate including Syncona, Oxford Science Enterprises, GV, Northpond Ventures, Sanofi Ventures and British Patient Capital.
To learn more, please visit www.omass.com. Follow us on LinkedIn.

Participants Who Complete Treatment in Global Phase 2b PRIZM MELAS Study Are Eligible to Enroll
CAMBRIDGE, Mass., August 20, 2025 – Tisento Therapeutics today announced that the first participant has enrolled in the company’s open-label extension study in MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Episodes). The extension study (NCT06961344) is evaluating the long- term safety and tolerability of zagociguat in individuals with MELAS who complete treatment in the global Phase 2b PRIZM study, which is expected to complete screening in the next months. The extension study is intended to provide uninterrupted access to zagociguat for clinical trial participants for up to two years and evaluate the long-term safety of zagociguat.
“We’re pleased to reach the important milestone of enrolling the first PRIZM study participant into our open-label extension study,” said Peter Hecht, Ph.D., chief executive officer of Tisento. “Enthusiastic engagement by MELAS patients and physicians is powering momentum in our development program, and we look forward to completing PRIZM enrollment in the next few months.”
The PRIZM study is actively enrolling in the U.S., Canada, Australia, United Kingdom, Italy, and Germany, and interested individuals are encouraged to discuss participation with their physician. PRIZM is evaluating the impact of zagociguat treatment on fatigue, cognitive performance, and other key aspects of MELAS. The clinical outcome assessments and endpoint strategy for the PRIZM study were informed by Tisento’s interview study in which individuals living with MELAS described the symptoms and impacts of the disease that are most important to them. Participants who complete treatment in PRIZM have the opportunity to enroll in the open-label extension study.
About the PRIZM Study
PRIZM – a Phase 2b Randomized, Placebo-Controlled Trial Investigating Zagociguat in MELAS – is evaluating the efficacy and safety of oral zagociguat 15 mg or 30 mg compared to placebo when administered once-daily for 12 weeks in participants with genetically and phenotypically defined MELAS. The PRIZM study has a crossover design, with two 12-week treatment periods separated by a 4- week washout period. All participants will receive zagociguat during one of the 12-week periods and placebo during the other. Participants who complete treatment in the study have the opportunity to enroll in an open-label extension study.
The global PRIZM study is now enrolling approximately 44 participants at mitochondrial disease centers of excellence in the U.S., Italy, Germany, United Kingdom, Australia, and Canada. For more information, please visit www.tisentotx.com/prizm or ClinicalTrials.gov (NCT06402123). Interested individuals can also reach out to their physicians for participation details.
About Zagociguat
Zagociguat is a once-daily, oral, clinical-stage investigational medicine with potential to positively impact both peripheral and central nervous system manifestations of mitochondrial diseases. Zagociguat stimulates soluble guanylate cyclase (sGC), an enzyme that is found in virtually every cell in every tissue of the body and is part of a system of cellular mechanisms that control critical physiological functions including neuronal function and blood flow.
A first-in-class, brain-penetrant sGC stimulator, zagociguat is hypothesized to rebalance dysregulated cellular pathways in MELAS. By restoring cellular functions that support mitochondria, zagociguat may help restore mitochondrial energy production and physiological function.
In a Phase 2a study in patients with MELAS, zagociguat exhibited a favorable safety profile, exposure throughout the body including in the central nervous system, and improvements in neuronal function, mitochondrial function, and blood flow in the brain. Zagociguat is currently being evaluated as a treatment for MELAS in the Phase 2b PRIZM study.
Zagociguat received Fast Track designation from the U.S. Food and Drug Administration for the treatment of MELAS. Fast Track is a process designed to facilitate the development and potentially expedite the review of medicines to treat serious conditions and fill an unmet medical need, with the goal of getting important new drugs to patients earlier.
For more information, visit www.tisentotx.com/our-science.
About Tisento Therapeutics
Tisento Therapeutics, a privately held biotech company, is developing novel medicines to treat diseases with significant unmet need, beginning with MELAS and other genetic mitochondrial diseases. Ti sento means “I hear you” in Italian; our approach to innovation begins with listening to patients and then channeling what we learn into decisive actions that shape our research and clinical programs.
Tisento is guided by a high-caliber internal team of biopharma veterans and an extensive external network of expert physicians, patient advocacy groups, researchers, industry-leading vendors, and other close collaborators who are partners in our mission to develop meaningful treatments for mitochondrial diseases.
Learn more at our website, www.tisentotx.com, or connect with us on LinkedIn, Facebook, X (@tisentotx), or Bluesky.
Contact
Tisento Media Relations
Jessi Rennekamp, Astrior Communications
Email: jessi@astriorcomms.com

-- Pioneering approach to halting the chronic neuroinflammation responsible for neurodegeneration
- Trial to assess safety, PK and detect early signs of efficacy
- Initial data expected in mid-2026
Sacramento, July 29, 2025 (GLOBE NEWSWIRE) - Therini Bio, Inc., a clinical-stage biotech company developing fibrin-targeting immunotherapies for neurodegenerative diseases driven by vascular dysfunction today announced that the first patient has been dosed in a Phase 1b trial evaluating THN391 for the treatment of Alzheimer's disease (AD). Therini’s novel approach aims to target chronic neuroinflammation responsible for neuronal loss in neurodegenerative disorders, including AD.
THN391 is a potential first-in-class, high-affinity, humanized monoclonal antibody designed to selectively target the inflammatory epitope on fibrin to treat inflammatory neurodegenerative diseases. THN391 demonstrated activity in preclinical studies in AD models in protecting against neuronal degeneration by blocking the activation of inflammatory cascades caused by toxic fibrin deposits. In a recent Phase 1a, randomized, double-blind, placebo-controlled, healthy volunteer trial, THN391 was shown to be well-tolerated, with no serious or drug related adverse events reported and had no adverse effects on coagulation. THN391 also exhibited dose proportional pharmacokinetics (PK) and a 40-day half-life that supports monthly dosing.
The Phase 1b randomized, double-blind, placebo-controlled trial will assess the safety, tolerability and PK of multiple ascending doses of THN391 in patients aged 65-85 years old with early AD and vascular risk factors. The trial will consist of at least three (3) dose cohorts with the patients in each cohort receiving three (3) monthly doses of THN391 or placebo and follow-up evaluation out to five (5) months. In addition to analyzing safety and PK data, biological and clinical endpoints will be assessed, utilizing pharmacodynamic disease biomarkers in plasma and cerebrospinal fluid, MRI imaging and cognitive assessments. Initial data is expected to be available in mid-2026.
Brad Navia, M.D., Ph.D., Chief Medical Officer – Neurology of Therini Bio, an internationally recognized expert in neuroscience research, clinical development and brain imaging, whose previous experience includes leading programs at Johnson & Johnson, Eisai Pharmaceuticals and Sunovion Pharmaceuticals, said, “THN391 represents a promising new approach to treating Alzheimer’s disease by directly addressing one of its primary causes, neuroinflammation. Its development also comes at an exciting time in the field as more research emerges on the important role of vascular dysfunction in AD. The flexible design of this Phase 1b trial enables us to engage an early AD population known to have vascular risk factors and employ innovative biomarkers aimed at detecting early signs of efficacy in the hopes of rapidly advancing this novel treatment towards providing relief for patients and families struggling with this dreadful disease.”
About
Therini Bio is a clinical-stage biotech company developing immunotherapies for neuroinflammation in diseases driven by vascular dysfunction. Therini Bio is developing a pipeline of potential first-in-class therapies selectively targeting toxic fibrin accumulation for diseases, including Alzheimer’s disease and Diabetic Macular Edema, where destructive neuroinflammation plays a central role in the disease process. Therini Bio’s top-tier syndicate of life sciences investors includes the Alzheimer’s Drug Discovery Foundation, Angelini Ventures, Apollo Health Ventures, SV Health Investors’ Biotech Fund and Dementia Discovery Fund, Dolby Family Ventures, Dreavent Biotech Investments, Eli Lilly and Company, Foundation for a Better World, MRL Ventures Fund, the therapeutics-focused corporate venture fund of Merck & Co., Inc., and Sanofi Ventures. For more information, visit www.therinibio.com.
Contact
Tara Nickerson, Ph.D.
Chief Executive Officer
tnickerson@therinibio.com
Oxford, United Kingdom – 6th August 2025 – OMass Therapeutics (‘OMass’ or ‘the Company’), a biotechnology company identifying medicines against highly validated target ecosystems such as membrane proteins or intracellular complexes, today announces the appointment of Carol A. Schafer as non-executive Director and Chair of the Audit Committee.
Carol has more than 25 years of experience in investment banking, equity capital markets, corporate finance and business development in the healthcare sector. She currently serves on the Board of Directors for Insmed Incorporated (Nasdaq: INSM), Immunome, Inc. (Nasdaq: IMNM), Kura Oncology, Inc (Nasdaq: KURA) and Repare Therapeutics Inc. (Nasdaq: RPTX). In addition, Carol previously served on the Board of Directors of Five Prime Therapeutics, Inc. (acquired by Amgen for $1.9 billion in April 2021) and Idera Pharmaceuticals, Inc. (until its merger in September 2022).
From 2007 to 2018, Carol worked at Wells Fargo Securities in a variety of roles, including as Vice Chair, Equity Capital Markets. Prior to this, Carol worked at J.P. Morgan, where she held positions of increasing responsibility including Managing Director, Equity Capital Markets. She holds a Master of Business Administration from New York University’s Stern School of Business and a Bachelor of Arts from Boston College.
“I am honoured to join the Board and contribute to the OMass’s mission of developing first or best-in-class small molecules for areas of high unmet need in endocrinology and immunology,” commented Carol Schafer, non-executive Director and Chair of the Audit Committee at OMass Therapeutics. “I look forward to working with the Board to help guide the company as they advance towards the clinic and prepare for their next phase of growth.”
“Carol’s extensive combination of healthcare investment banking experience and biotech board experience will be invaluable to OMass as we move towards becoming a clinical-stage company,” added Jim Geraghty, Chairman of OMass Therapeutics’ Board of Directors. “I am confident that her strategic insight and understanding of equity markets will be a tremendous asset to the company. We are pleased to welcome her to the Board and as our new audit committee chair.”
-ENDS-
For further information, please contact:
|
OMass Therapeutics |
|
|
Rosamond Deegan, Chief Executive Officer Email: ros.deegan@omass.com |
|
|
ICR Healthcare (For media) |
|
|
Namrata Taak / Ashley Tapp Email: omass@icrhealthcare.com |
|
|
Thrust Strategic Communications (For IR) Renee Leck Email: renee@thrustsc.com |
|
About OMass Therapeutics
OMass Therapeutics is a biotechnology company discovering medicines against highly-validated target ecosystems, such as membrane proteins or intracellular complexes.
OdyssION™, OMass’ unique drug discovery platform, comprises next-generation native mass spectrometry with novel biochemistry techniques and custom chemistry to interrogate not just a drug target, but also the interaction of the target with its native ecosystem, separate from the confounding complexity of the cell. This unique approach results in cell-system fidelity with cell-free precision.
OMass is advancing a pipeline of small molecule therapeutics in rare diseases and immunological conditions. Its lead program is a best-in-class MC2 (melanocortin-2) receptor antagonist for the treatment of Congenital Adrenal Hyperplasia (CAH) and ACTH-dependent Cushing syndrome. The focus of the program has been to increase receptor residency time to make OMass’ antagonists resistant to competition by the endogenous ligand, thereby avoiding loss of efficacy in the face of rising adrenocorticotropic hormone (ACTH) levels due to reductions in glucocorticoid supplementation for CAH or progression of Cushing’s Syndrome.
Headquartered in Oxford, UK, OMass has raised over $160M (£129M) from a top-tier international investor syndicate including Syncona, Oxford Science Enterprises, GV, Northpond Ventures, Sanofi Ventures and British Patient Capital.
To learn more, please visit www.omass.com. Follow us on LinkedIn.
- Former Amgen, Novo Nordisk and Boehringer Ingelheim senior leader joins NodThera Leadership Team
- Will spearhead clinical advancement of the company’s innovative NLRP3 inflammasome inhibitor portfolio
- Brings deep clinical leadership in obesity and cardiometabolic diseases, with a proven track record guiding programs from early-stage development through Phase 3 and regulatory approval
Philadelphia, PA, July 21, 2025 - NodThera, a leading clinical-stage biotech delivering a paradigm shift in the treatment of chronic inflammatory diseases through selective modulation of the NLRP3 inflammasome, today announces the appointment of Dr. Jyothis George, M.D., Ph.D., FRCP, FACE as Chief Medical Officer, effective immediately.
Jyothis brings a wealth of global leadership experience in cardiometabolic drug development, regulatory strategy and medical affairs. He joins NodThera from Amgen (NASDAQ: AMGN), where he served as Global Medical Vice President, Obesity and Related Conditions, driving strategic decision making on target indications and priority patient populations, securing investment to generate differentiating evidence and leading expert engagement. His recent responsibilities included strategic planning around MariTide, Amgen’s lead candidate in obesity.
Previously, Jyothis held leadership roles at Novo Nordisk (CPH: NOVO-B) and Boehringer Ingelheim, directing pivotal programs in obesity, diabetes, heart failure and chronic kidney disease. At Novo Nordisk, Jyothis was Corporate Vice President, Clinical Development, Medical Affairs and Regulatory. There he successfully delivered multiple key clinical trials across the Novo Nordisk portfolio, including major regulatory studies for several assets including semaglutide (Wegovy®) and launching impactful educational platforms such as ObesityME and the Diabetes Masters Network. At Boehringer Ingelheim, as Global Head of Clinical Development and Medical Affairs, Cardiometabolism, he led the clinical development and medical affairs teams for two blockbuster cardiometabolic assets: empagliflozin, the first diabetes treatment to gain an FDA cardiovascular mortality indication, and the DPP4 inhibitor linagliptin. During his tenure, he successfully delivered multiple global clinical trials, including serving on steering committees of six major cardiovascular outcomes trials.
Daniel Swisher, Chief Executive Officer of NodThera, said: "We are thrilled to welcome Jyothis to NodThera as our Chief Medical Officer. He brings a critical combination of deep clinical and patient insight coupled to recent global leadership in cardiometabolic drug development, shaping landmark trials and delivering regulatory success. His expertise will be invaluable to NodThera as we advance our brainpenetrant NLRP3 inflammasome inhibitors further through clinical development."
Jyothis George, M.D., Ph.D., FRCP, FACE, commented: "Joining NodThera provides an exciting opportunity to transform the treatment of diseases driven by chronic low-grade inflammation. I am inspired by the science and the team’s bold vision to reimagine what’s possible for patients by restoring the body’s natural metabolic balance. Together with the team, I am looking forward to building on NodThera’s promising clinical momentum to deliver therapies that are not only differentiated, but truly transformative for patients."
Jyothis holds an M.B.B.S. from St. John’s Medical College in Bangalore, India; a Ph.D. in Neuroendocrinology from the University of Edinburgh, and is board certified in Internal Medicine, Endocrinology and Diabetes (UK). He is a Fellow of the Royal College of Physicians (Edinburgh) and the American College of Endocrinology and has authored more than 100 peer-reviewed publications. He has previously served on the boards of Amgen Technologies Ireland and Oxford Centre for Diabetes, Endocrinology and Metabolism.
For more information about NodThera please contact:
NodThera
Tel: +44 (0) 1223 608130
Email: info@nodthera.com
ICR Healthcare
Amber Fennell, David Daley
Tel: +44 (0)20 3709 5700
Email: nodthera@icrhealthcare.com
About NodThera
NodThera is a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases. Led by an experienced management team, NodThera is combining a deep understanding of NLRP3 inhibition, pharmaceutical neuroscience expertise and precision chemistry. Its two lead clinical candidates are oral, small molecule NLRP3 inflammasome inhibitors, which have demonstrated differentiated, potentially best-in-class clinical profiles with significant antiinflammatory effects and high brain penetration, offering distinct opportunities to treat multiple indications. The Company is backed by top-tier investors including 5AM Ventures, Blue Owl Capital, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Philadelphia, Pennsylvania, with additional operations in Cambridge, UK. Learn more at www.nodthera.com or follow the Company on LinkedIn.
- Pioneering approach to halting the chronic neuroinflammation responsible for neurodegeneration
- Trial to assess safety, PK and detect early signs of efficacy
- Initial data expected in mid-2026
Sacramento, July 29, 2025 (GLOBE NEWSWIRE) - Therini Bio, Inc., a clinical-stage biotech company developing fibrin-targeting immunotherapies for neurodegenerative diseases driven by vascular dysfunction today announced that the first patient has been dosed in a Phase 1b trial evaluating THN391 for the treatment of Alzheimer's disease (AD). Therini’s novel approach aims to target chronic neuroinflammation responsible for neuronal loss in neurodegenerative disorders, including AD.
THN391 is a potential first-in-class, high-affinity, humanized monoclonal antibody designed to selectively target the inflammatory epitope on fibrin to treat inflammatory neurodegenerative diseases. THN391 demonstrated activity in preclinical studies in AD models in protecting against neuronal degeneration by blocking the activation of inflammatory cascades caused by toxic fibrin deposits. In a recent Phase 1a, randomized, double-blind, placebo-controlled, healthy volunteer trial, THN391 was shown to be well-tolerated, with no serious or drug related adverse events reported and had no adverse effects on coagulation. THN391 also exhibited dose proportional pharmacokinetics (PK) and a 40-day half-life that supports monthly dosing.
The Phase 1b randomized, double-blind, placebo-controlled trial will assess the safety, tolerability and PK of multiple ascending doses of THN391 in patients aged 65-85 years old with early AD and vascular risk factors. The trial will consist of at least three (3) dose cohorts with the patients in each cohort receiving three (3) monthly doses of THN391 or placebo and follow-up evaluation out to five (5) months. In addition to analyzing safety and PK data, biological and clinical endpoints will be assessed, utilizing pharmacodynamic disease biomarkers in plasma and cerebrospinal fluid, MRI imaging and cognitive assessments. Initial data is expected to be available in mid-2026.
Brad Navia, M.D., Ph.D., Chief Medical Officer – Neurology of Therini Bio, an internationally recognized expert in neuroscience research, clinical development and brain imaging, whose previous experience includes leading programs at Johnson & Johnson, Eisai Pharmaceuticals and Sunovion Pharmaceuticals, said, “THN391 represents a promising new approach to treating Alzheimer’s disease by directly addressing one of its primary causes, neuroinflammation. Its development also comes at an exciting time in the field as more research emerges on the important role of vascular dysfunction in AD. The flexible design of this Phase 1b trial enables us to engage an early AD population known to have vascular risk factors and employ innovative biomarkers aimed at detecting early signs of efficacy in the hopes of rapidly advancing this novel treatment towards providing relief for patients and families struggling with this dreadful disease.”
About
Therini Bio is a clinical-stage biotech company developing immunotherapies for neuroinflammation in diseases driven by vascular dysfunction. Therini Bio is developing a pipeline of potential first-in-class therapies selectively targeting toxic fibrin accumulation for diseases, including Alzheimer’s disease and Diabetic Macular Edema, where destructive neuroinflammation plays a central role in the disease process. Therini Bio’s top-tier syndicate of life sciences investors includes the Alzheimer’s Drug Discovery Foundation, Angelini Ventures, Apollo Health Ventures, SV Health Investors’ Biotech Fund and Dementia Discovery Fund, Dolby Family Ventures, Dreavent Biotech Investments, Eli Lilly and Company, Foundation for a Better World, MRL Ventures Fund, the therapeutics-focused corporate venture fund of Merck & Co., Inc., and Sanofi Ventures. For more information, visit www.therinibio.com.
Contact
Tara Nickerson, Ph.D.
Chief Executive Officer
tnickerson@therinibio.com

- Matchpoint to receive up to $60 million in upfront payment and research funding, with up to $1 billion in total potential payments, including option exercise fee, development and commercial milestones
- Matchpoint will lead all research activities through development candidate selection
Watertown, MA – July 24, 2025 – Matchpoint Therapeutics, a biotechnology company pioneering the discovery of precision covalent medicines, today announced that it has entered into an exclusive option and license agreement with Novartis for the development and commercialization of oral covalent inhibitors directed at a transcription factor linked to a number of inflammatory diseases.
“This collaboration highlights the potential of our targeted approach to covalent chemistry to unlock novel mechanisms and achieve superior pharmacology,” said Andre Turenne, President and CEO of Matchpoint Therapeutics. “We are thrilled to collaborate with Novartis, not only for its leadership in immunology research, but also for its outstanding drug development and commercialization track record. These capabilities will ensure that our program has the maximum impact for patients.”
Matchpoint’s approach leverages the properties of covalent chemistry and a proprietary platform to target a novel binding site on a historically hard-to-drug protein. The result is to precisely inhibit that protein’s function and the resulting production of pro-inflammatory cytokines and chemokines.
"Novartis is deeply committed to pioneering innovative approaches for patients living with inflammatory and autoimmune diseases,” said Richard Siegel, M.D., Ph.D., Global Head of Immunology Research at Novartis. “We are excited to partner with Matchpoint to advance covalent medicines with the potential to address unmet medical needs and historically undruggable targets."
Under the terms of the agreement, Matchpoint will utilize its Advanced Covalent Exploration (ACE)TM platform to advance its preclinical program through development candidate selection. Matchpoint will lead all research activities to that point and will receive research funding from Novartis. If Novartis exercises its option to exclusively license the program, Novartis will have global rights to develop and commercialize all products resulting from the collaboration. Matchpoint is eligible to receive up to $1 billion in total payments, including development and commercial milestones, plus tiered royalties on future product sales.
Aquilo Partners LP and Ropes & Gray LLP acted as financial and legal advisors to Matchpoint on this transaction, respectively.
About Matchpoint Therapeutics
Matchpoint is a privately held biotechnology company harnessing the power of covalency to discover precision oral medicines to transform the treatment of immune diseases. The company’s Advanced Covalent Exploration (ACE)TM platform integrates a suite of leading covalent drug discovery capabilities and is deployed toward validated, disease-causing protein targets that are yet undrugged or inadequately addressed by conventional approaches. Matchpoint is based in Watertown, MA. For more information, visit www.matchpointtx.com.
Media Contact:
Cory Tromblee
ScientPR
Media@MatchpointTx.com
- OMS1620 is advancing through IND-enabling studies for diseases associated with ACTH excess, including congenital adrenal hyperplasia
- Designed to resist competition from endogenous ACTH by maximizing receptor residency time
- OMS1620 demonstrates best-in-class efficacy in acute and chronic preclinical models of ACTH excess
Oxford, United Kingdom – 14th July 2025 – OMass Therapeutics (‘OMass’ or ‘the Company’), a biotechnology company identifying medicines against highly validated target ecosystems such as membrane proteins or intracellular complexes, today announces positive preclinical data for OMS1620, its lead program targeting the melanocortin-2 (MC2) receptor, at ENDO 2025, the Annual Endocrine Society Meeting, taking place in San Francisco from 12-15 July.
This poster is the first public data disclosure related to OMS1620, a potential best-in-class MC2R antagonist currently in IND-enabling studies. MC2R is a GPCR for the adrenocorticotropic hormone (ACTH), a hormone released by the pituitary that triggers cortisol and androgen production. OMS1620 is being developed for diseases associated with ACTH excess, including congenital adrenal hyperplasia (CAH).
In classical CAH, patients are unable to produce cortisol and require exogenous glucocorticoid supplementation. In people without CAH, endogenous cortisol prevents ACTH upregulation but to achieve this in CAH patients, supraphysiological doses of glucocorticoids are usually required. This results in CAH patients having symptoms associated with ACTH over production (leading to androgen excess), side effects associated with glucocorticoid overdosing, or both.
ACTH surges in CAH can be many times above the upper limit of normal, last for multiple hours, and increase as patients try to downtitrate their glucocorticoid dose. OMS1620 has been exquisitely designed to maximize receptor residency time, making it highly resistant to competition from rising endogenous ACTH. This was demonstrated preclinically utilizing an acute ACTH challenge model in rats, an experiment meant to mimic the high levels of ACTH observed in CAH patients. Compounds with longer residence time were shown to have greater MC2 receptor inhibition. OMS1620 also led to improvement in body and adrenal weight in a chronic ACTH excess model, where Sprague Dawley rats were treated with ACTH via an osmotic mini-pump.
These results demonstrate OMS1620 is a potential best-in-class MC2 antagonist with the ideal compound properties to combat high ACTH surges that CAH patients can experience throughout the day. This can further support patients in achieving the ultimate treatment goal in CAH of androgen normalization whilst on physiological dose replacement of glucocorticoids.
Ros Deegan, Chief Executive Officer at OMass Therapeutics, commented: “This compelling preclinical data validates our approach of optimizing our MC2 antagonists’ residence time to deliver a best-in-class molecule in OMS1620. Our preclinical modelling suggests OMS1620’s profile can further support patients as they try to achieve the ultimate treatment goal in CAH of normalizing androgen levels while on physiological doses of glucocorticoids. This underscores the potential of our MC2 program to transform outcomes for patients with CAH, a population with significant unmet need and we’re excited to take the next step towards the clinic.”
The poster can be downloaded here.
-ENDS-
For further information, please contact:
|
OMass Therapeutics |
ICR Healthcare |
|
Rosamond Deegan, Chief Executive Officer Email: ros.deegan@omass.com |
Namrata Taak / Ashley Tapp Email: omass@icrhealthcare.com |
About OMass Therapeutics
OMass Therapeutics is a biotechnology company discovering medicines against highly-validated target ecosystems, such as membrane proteins or intracellular complexes.
OdyssION™, OMass’ unique drug discovery platform, comprises next-generation native mass spectrometry with novel biochemistry techniques and custom chemistry to interrogate not just a drug target, but also the interaction of the target with its native ecosystem, separate from the confounding complexity of the cell. This unique approach results in cell-system fidelity with cell-free precision.
OMass is advancing a pipeline of small molecule therapeutics in rare diseases and immunological conditions. Its lead program is a best-in-class MC2 (melanocortin-2) receptor antagonist for the treatment of Congenital Adrenal Hyperplasia (CAH) and ACTH-dependent Cushing syndrome. The focus of the program has been to increase receptor residency time to make OMass’ antagonists resistant to competition by the endogenous ligand, thereby avoiding loss of efficacy in the face of rising adrenocorticotropic hormone (ACTH) levels due to reductions in glucocorticoid supplementation for CAH or progression of Cushing’s Syndrome.
Headquartered in Oxford, UK, OMass has raised over $160M (£129M) from a top-tier international investor syndicate including Syncona, Oxford Science Enterprises, GV, Northpond Ventures, Sanofi Ventures and British Patient Capital.
To learn more, please visit www.omass.com. Follow us on LinkedIn.
- Strong track record at executive and board level of building company value across all stages of development including supporting the development of multiple blockbuster medicines
Cardiff, United Kingdom – 15 July 2025 – Draig Therapeutics (“Draig”), a clinical-stage company aiming to transform the treatment of neuropsychiatric diseases, today announces the appointment of Douglas E. Williams, Ph.D. as independent Chair of its Board of Directors. He takes over from Ruth McKernan, who retains her role on the Board as a Director and Interim Chief Executive Officer (CEO).
Dr Williams brings more than 30 years of experience in the biopharmaceutical sector, having held executive leadership and board roles across some of the industry’s most innovative companies including Biogen, ZymoGenetics, Amgen and Seattle Genetics and contributed to the development of several blockbusters therapies, including Enbrel®, Tecfidera® and Spinraza®.
Ruth McKernan, Interim CEO and founder of Draig Therapeutics, said, “Douglas’s exceptional track record of developing innovative therapies and driving strategic growth makes him an ideal leader for our Board. I could not be more delighted. The Company looks forward to benefitting from his leadership and guidance in delivering transformative neuropsychiatric therapies.”
“I am honoured to take on the role of Chair as Draig Therapeutics advances its exciting pipeline into the next phases of clinical development,” said Douglas E. Williams, incoming Chair of Draig Therapeutics. “More effective therapies for major depressive disorder and other neuropsychiatric disorders remain significant unmet needs for millions of people worldwide. Draig’s scientific approach to develop novel medicines for these patients is both compelling and differentiated, and I look forward to working closely with the board and its exceptional leadership team to support the Company achieve its goals.”
Most recently, Dr Williams served as the President of R&D at Sana Biotechnology, a leading cell therapy company. Prior to joining Sana, he was the founding CEO of Codiak Biosciences, where he led the company through several private financing rounds as well as a successful initial public offering. Previously, Dr Williams was Executive Vice President (EVP) of Research and Development at Biogen where he played a key role in advancing multiple programmes. Before joining Biogen, he was CEO and a board member at ZymoGenetics, where he led the company through its acquisition by Bristol Myers Squibb in 2010.
Earlier in his career, Dr Williams held senior leadership roles at several prominent biotech companies, including Chief Scientific Officer and EVP R&D at Seattle Genetics (Seagen, acquired by Pfizer in 2023 for $43 billion), and Senior VP and Washington Site Leader at Amgen. He spent over a decade at Immunex, where he served as EVP and Chief Technology Officer, and was a member of the Board of Directors.
Dr Williams currently serves on the boards of numerous biotechnology companies, including Climb Bio (Chair) and CAMP4 Therapeutics, eGenesis and Stablix (Director). He has also previously been Chair or Director on the boards of multiple private and public companies in the US and Europe over the past 30 years, including CytoImmune Therapeutics, Xenikos, Array Biopharma, AC Immune, Ovid Therapeutics, Ironwood Pharmaceuticals and Regulus Therapeutics, among others.
ENDS
For further information:
Mark Swallow, Sandi Greenwood
E-mail: draigtx@medistrava.com
Draig Therapeutics
E-mail: rmckernan@draigtherapeutics.com
About Draig Therapeutics
Draig Therapeutics is a clinical-stage company with a mission to transform treatments in Neuropsychiatry. The company is leveraging its founders’ unique scientific expertise in modulating the core glutamate / GABA pathways that play a critical role in neuropsychiatric diseases to advance a pipeline of groundbreaking therapies designed to address large unmet patient needs, including in Major Depressive Disorder (MDD).
Draig is the Welsh word for ‘dragon’ and it reflects the company’s origins in Wales. The name and logo were inspired by this heritage, reflecting its scientific roots stemming from Cardiff University.
Draig was co-founded by Cardiff University and SV Health Investors, which led the seed financing with ICG, and is backed by other leading healthcare venture firms including Access Biotechnology, Canaan Partners, SR One, Sanofi Ventures and Schroders Capital. For more information, please visit www.draigtherapeutics.com
Oxford, United Kingdom – 7th July 2025 – OMass Therapeutics (‘OMass’ or ‘the Company’), a biotechnology company identifying medicines against highly validated target ecosystems such as membrane proteins or intracellular complexes, today announces that preclinical data for OMS1620, it’s lead program targeting the melanocortin-2 (MC2) receptor, will be presented at ENDO 2025, the Annual Endocrine Society Meeting, taking place in San Francisco from 12-15 July.
This poster will be the first public data disclosure related to OMass’ MC2 receptor program and OMS1620, its development candidate currently in IND enabling studies. MC2R is a GPCR for the adrenocorticotropic hormone (ACTH), which is a hormone released by the pituitary that triggers cortisol and androgen production. OMS1620 is a potential best-in-class MC2 antagonist for diseases associated with ACTH excess, including congenital adrenal hyperplasia (CAH).
In classical CAH, patients are unable to produce cortisol, leading to the chronic overproduction of ACTH which drives excess androgen production. Due to the lack of cortisol, CAH patients must receive glucocorticoid supplementation to be able to survive. In people without CAH, endogenous cortisol prevents ACTH upregulation but to achieve this in CAH patients, supraphysiological doses of glucocorticoids are usually required. This results in patients either suffering from effects of hyperandrogenism, over-dosing of glucocorticoids, or both.
OMS1620 has been exquisitely designed to maximize receptor residency time, making it highly resistant to competition from rising endogenous ACTH that occur as glucocorticoid doses are reduced. This can allow patients to achieve the ultimate treatment goal in CAH of androgen normalization whilst on physiological dose replacement of glucocorticoids.
Poster Details:
Title: Optimizing Binding Kinetics to Develop Insurmountable MC2 Receptor Antagonists for the Treatment of Congenital Adrenal Hyperplasia
Presenter: Mark Soave
Date/Time: 13 July 2025/12-1.30PM PDT
Session: Session P55 - ADRENAL (EXCLUDING MINERALOCORTICOIDS): Adrenal Insufficiency and CAH II
Location: Poster Area, Moscone Convention Centre, San Francisco
Ros Deegan, Chief Executive Officer of OMass, said: “We are excited to share the preclinical data we have generated for OMS1620 at ENDO 2025 as we continue our progress towards the clinic. We believe that OMS1620 has the potential to be a best-in-class product with a differentiated profile, that can ultimately improve the lives of patients with diseases associated with ACTH excess”.
-ENDS-
For further information, please contact:
|
OMass Therapeutics |
ICR Healthcare |
|
Rosamond Deegan, Chief Executive Officer Phone: +44 (0)1235 527589 Email: ros.deegan@omass.com |
Namrata Taak / Ashley Tapp Phone: +44 (0)20 3709 5700 Email: omass@icrhealthcare.com |
About OMass Therapeutics
OMass Therapeutics is a biotechnology company discovering medicines against highly-validated target ecosystems, such as membrane proteins or intracellular complexes.
OdyssION™, OMass’ unique drug discovery platform, comprises next-generation native mass spectrometry with novel biochemistry techniques and custom chemistry to interrogate not just a drug target, but also the interaction of the target with its native ecosystem, separate from the confounding complexity of the cell. This unique approach results in cell-system fidelity with cell-free precision.
OMass is advancing a pipeline of small molecule therapeutics in rare diseases and immunological conditions. Its lead program is a best-in-class MC2 (melanocortin-2) receptor antagonist for the treatment of Congenital Adrenal Hyperplasia (CAH) and ACTH-dependent Cushing syndrome. The focus of the program has been to increase receptor residency time to make OMass’ antagonists resistant to competition by the endogenous ligand, thereby avoiding loss of efficacy in the face of rising adrenocorticotropic hormone (ACTH) levels due to reductions in glucocorticoid supplementation for CAH or progression of Cushing’s Syndrome.
Headquartered in Oxford, UK, OMass has raised over $160M (£129M) from a top-tier international investor syndicate including Syncona, Oxford Science Enterprises, GV, Northpond Ventures, Sanofi Ventures and British Patient Capital.
To learn more, please visit www.omass.com. Follow us on LinkedIn.

- Joanne Kotz joins Atalanta as Chief Executive Officer, with proven experience building and scaling innovative platform-based companies
- Doug Pagán joins as Chief Financial Officer & Chief Operating Officer, bringing deep financial, business strategy and RNAi expertise
BOSTON, June 18, 2025 – Atalanta Therapeutics, a biotechnology company pioneering RNA interference (RNAi) for the treatment of intractable neurological diseases, announced today that Joanne Kotz has been named Chief Executive Officer and a member of the Board of Directors, succeeding Alicia Secor, who has stepped down as part of a planned transition to pursue other opportunities. The company also announced the appointment of Douglas Pagán as Chief Financial Officer & Chief Operating Officer, succeeding Jeffrey Young.
Additionally, Bob D. Brown, an industry veteran with extensive RNAi drug discovery and development experience, has joined the company’s Scientific Advisory Board to advise as the company advances its innovative pipeline into the clinic.
"I want to thank Alicia for her extraordinary leadership of the company from formation to the cusp of entry into the clinic, as well as her continued role as an advisor through this transition,” said Dr. Stephen Knight, President and Senior Managing Partner of F-Prime and a member of the Atalanta Board of Directors. “We are delighted to have Joanne lead Atalanta’s next stage. Joanne is an accomplished leader who has demonstrated the ability to advance innovative platforms and pipelines through the transition to clinical stage, and has optimized company value creation through strong financings, partnerships and strategic transactions.”
“I’m thrilled to be joining Atalanta, which is pioneering the effort to bring RNAi therapies to patients living with devastating neurological diseases,” said Dr. Kotz. “I am passionately committed to Atalanta’s mission to deliver life-transforming therapies to patients and look forward to working with the team to transition our first programs into the clinic as we work to realize the full therapeutic potential of Atalanta’s innovative di-siRNA platform.”
“Over the past six years we’ve made incredible scientific progress at Atalanta. I’m proud of all that we’ve been able to accomplish together and thank the Board and team for their support and dedication to our mission,” said Ms. Secor. “The transition from research to clinical stage is a critical time for any biotech, and Joanne’s leadership and experience is ideally suited to guide Atalanta’s continued progress towards our mission of delivering RNAi medicines to patients living with severe neurological diseases.”
Dr. Kotz brings to Atalanta extensive executive experience, including most recently as cofounder and CEO of Jnana Therapeutics. She led the company from an early discovery platform stage through the transition to clinical stage. Jnana was acquired by Otsuka Pharmaceutical in 2024 for ~$1 billion following the demonstration of positive clinical proof of concept for the company’s lead rare disease program. Prior to Jnana, Dr. Kotz held leadership roles at FBRI, F-Prime Capital’s initiative to enable therapeutic breakthroughs in Alzheimer’s disease and related brain disorders, and at the Broad Institute. She is a member of the Board of Directors of the Chordoma Foundation. Dr. Kotz received her Ph.D. in chemistry from the University of California, Berkeley and a B.S. in chemistry from the University of Florida. She conducted postdoctoral research at Genentech and at the Necker Children’s Hospital in Paris.
Mr. Pagán brings to Atalanta more than two decades of experience in finance, investor relations, and capital formation across both public and venture-backed biopharmaceutical companies. Prior to joining Atalanta, he served as Chief Financial Officer and Chief Operating Officer at Jnana Therapeutics, where he was instrumental in securing the company’s $107 million Series C financing heading into the clinic, and the subsequent ~$1 billion acquisition by Otsuka Pharmaceutical. Prior to Jnana, Mr. Pagán was Chief Financial Officer at the RNAi company Dicerna Pharmaceuticals, where he oversaw the 2021 sale of the company to Novo Nordisk for $3.3 billion. He has previously held leadership roles at Acceleron and Biogen and has served on the Board of Directors of the biotech companies Ziopharm Oncology and Timberlyne Therapeutics. Mr. Pagán holds an MBA from Columbia Business School and a BSE in chemical engineering from Princeton University.
Dr. Brown brings more than 30 years of experience in RNAi and oligonucleotide research and development, including from discovery through drug approval and across a wide range of clinical indications and regulatory jurisdictions. Most recently, Dr. Brown served as Chief Scientific Officer and Executive Vice President of R&D at Dicerna Pharmaceuticals, an RNAi-focused therapeutics company that was acquired by Novo
Nordisk. At Novo, he served as President and Head of the Dicerna Transformation Research Unit and SVP. Prior to Dicerna, Dr. Brown held various R&D leadership positions at Genta, a clinical-stage antisense oligonucleotide therapeutics company, and previously was a co- founder of Oasis Biosciences, which was acquired by Gen-Probe. Dr. Brown earned a Ph.D. in molecular biology from the University of California, Berkeley and B.S. degrees in chemistry and biology from the University of Washington, Seattle.
About Atalanta Therapeutics
Atalanta Therapeutics is a biotechnology company developing treatments for intractable diseases of the central nervous system using RNA interference. Atalanta’s unique platform of divalent small interfering RNA (di-siRNA) is designed to enable durable, selective gene silencing throughout the brain and spinal cord. Atalanta is advancing a wholly owned pipeline of disease-modifying programs for Huntington’s disease, genetic epilepsy, severe chronic pain, and other neurological diseases in addition to partnered programs as part of a strategic collaboration with Genentech. Atalanta is headquartered in Boston, Mass. For more information, visit www.atalantatx.com.
Media Contact:
Lisa Raffensperger
Ten Bridge Communications
lisa@tenbridgecommunications.com
- Former SVP at AstraZeneca R&D brings deep expertise in obesity and cardiometabolic research, portfolio strategy development and commercialization
- Appointment follows commencement of Phase 2 RESOLVE-1 trial of oral NLRP3 inflammasome inhibitor NT-0796 in patients with obesity
Philadelphia, PA, June 17, 2025 - NodThera, a leading clinical-stage biotech delivering a paradigm shift in the treatment of chronic inflammatory diseases through selective modulation of the NLRP3 inflammasome, today announces the appointment of Elisabeth Björk, M.D., Ph.D. as Board Member.
Elisabeth has more than 20 years of experience in late-stage clinical development, global regulatory submissions and successful drug commercialization, with particular strengths in cardiovascular and metabolic disease. She most recently served as Senior Vice President (SVP), Head of Obesity franchise at AstraZeneca (LON: AZN), where she led the development of AZD5004, a small molecule oral GLP-1 receptor agonist, and drove portfolio strategy development for obesity, weight management, diabetes, and cardiorenal protection.
While at AstraZeneca, Elisabeth also held senior CVRM (Cardiovascular, Renal and Metabolism) leadership roles including SVP, Head of late phase CVRM R&D. In this role she was instrumental in transforming the disease area from the treatment of individual risk factors to broad cardiorenal protection, bringing several blockbuster drugs to patients. Elisabeth also co-chaired the Therapy Area Leadership Team setting strategy across research and commercial functions, delivering global latestage portfolio and life cycle management programs, and serving as global line leader for CVRM clinical development personnel. Her leadership was pivotal in building AstraZeneca's world-class CVRM portfolio and establishing one of the industry's leading R&D sites in Gothenburg, Sweden, where she served as site lead.
Daniel Swisher, Chief Executive Officer of NodThera, said: "We are thrilled to welcome Elisabeth to our Board of Directors. Her exceptional track record in the obesity and broader cardiometabolic fields, setting and executing strategy, and bringing transformative medicines to patients, will be invaluable as we advance our brain-penetrant NLRP3 inflammasome inhibitors further through clinical development. Elisabeth's deep scientific and commercial understanding, in a therapeutic focus area that is so important to NodThera, will bring tremendous value to the advancement of our programs and our evolution into a mature, high-value clinical-stage company."
Elisabeth Björk, MD, PhD, commented: "I am delighted to join NodThera's Board of Directors at this critical point in the company's journey. The potential to selectively modulate the NLRP3 inflammasome to reset the body’s metabolic pathways represents a compelling therapeutic opportunity with significant implications for patients suffering from chronic inflammatory diseases. With my drug development experience, I look forward to supporting the talented and experienced management team as they advance their innovative therapies to market."
Elisabeth currently serves on the Boards of several public and private companies, including Rocket Pharmaceuticals (NASDAQ: RCKT), Pharvaris N.V. (NASDAQ: PHVS), Vicore Pharma AB (STO: VICO), Camurus AB (STO: CAMX), and Ousia Pharma ApS. Elisabeth holds an M.D. from the Karolinska Institute and a Ph.D. in Endocrinology from Uppsala University, where she also served as Associate Professor of Medicine.
For more information about NodThera please contact:
NodThera
Tel: +44 (0) 1223 608130
Email: info@nodthera.com
ICR Healthcare
Amber Fennell, David Daley
Tel: +44 (0)20 3709 5700
Email: nodthera@icrhealthcare.com
About NodThera
NodThera is a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases. Led by an experienced management team, NodThera is combining a deep understanding of NLRP3 inhibition, pharmaceutical neuroscience expertise and precision chemistry. Its two lead clinical candidates are oral, small molecule NLRP3 inflammasome inhibitors, which have demonstrated differentiated, potentially best-in-class clinical profiles with significant anti-inflammatory effects and high brain penetration, offering distinct opportunities to treat multiple indications. The Company is backed by top-tier investors including 5AM Ventures, Blue Owl Capital, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Philadelphia, Pennsylvania, with additional operations in Cambridge, UK. Learn more at www.nodthera.com or follow the Company on LinkedIn.
- Clinical-stage company led by world-class team targeting core neurotransmission pathways underlying neuropsychiatric disorders with a rich pipeline of novel, potentially transformative treatments
- Proceeds will fund four clinical studies: two related to the Phase 2-stage lead candidate, DT-101, in Major Depressive Disorder and two in additional programmes
- Outstanding syndicate of Series A investors led by Access Biotechnology join seed investors SV Health Investors and ICG
Cardiff, United Kingdom – 18 June 2025 – Draig Therapeutics (“Draig”), a clinical-stage company aiming to transform the treatment of neuropsychiatric diseases, today launches from stealth having raised a total of $140 million (£107 million) in the last nine months. The oversubscribed Series A financing was led by Access Biotechnology, with participation from Canaan Partners, SR One, Sanofi Ventures, Schroders Capital along with seed investors SV Health Investors, which co-founded the company, and ICG.
“Despite numerous treatments available for neuropsychiatric disorders, a significant unmet need remains with many patients continuing to experience inadequate symptom relief and high rates of relapse,” said Liam Ratcliffe, Head of Access Biotechnology. “Draig’s differentiated approach, which targets core mechanisms underlying these complex conditions, has the potential to deliver a real breakthrough for patients.”
Draig was formed through a partnership between Cardiff University’s Medicine Discovery Institute and SV Health Investors and launched in 2024. SV Health Investors and ICG provided initial seed funding and built the company.
The unique scientific expertise of Professors John Atack and Simon Ward in safely and effectively modulating the core glutamate and GABA pathways in the brain underpins Draig’s pipeline of novel drug candidates. Both founders are Directors of the Medicine Discovery Institute (MDI) at Cardiff University and have attracted a world-class leadership team of highly experienced industry experts and proven company builders with strong track records of driving innovation and developing transformative therapies into Draig.
“Making the best molecules to rebalance brain networks has been John and Simon’s life work. It has been a professional highlight for me to be part of creating this hugely promising company too,” said Ruth McKernan, Venture Partner at SV Health Investors, Co-Founder and Executive Chair of Draig.
“From the outset, we were drawn to Draig’s bold scientific vision and the founding team’s deep expertise in unlocking high value but previously difficult-to-drug targets in neuropsychiatric
disorders,” said Charles Dunn, Principal at SV Health Investors. “Built around an exciting clinical-stage asset, Draig is a perfect example of SV’s strategy to create and build world-class companies around innovative science to deliver high-impact drugs to patients and address major unmet needs.”
The new funds will enable Draig to advance its lead candidate DT-101, a next-generation AMPA receptor positive allosteric modulator (PAM), into Phase 2 trials for Major Depressive Disorder in 2025. DT-101 was designed to allow effective modulation of the AMPA receptor without compromising safety. This was borne out in data from a well-tolerated Phase 1a programme in over 60 subjects, which clearly demonstrated target engagement using the novel technique of magnetoencephalography and will be presented at an upcoming scientific meeting.
“With several exciting candidates in our pipeline, including our promising clinical candidate DT-101 for Major Depressive Disorder, alongside the support of an exceptional investor syndicate and a world-class team, Draig has a great platform from which to make a positive impact on the treatment of neuropsychiatric disorders,” said Samantha Budd Haeberlein, Venture Partner at ICG and former Chief Medical Officer of Draig.
The funding will also enable Draig to advance two highly selective GABAA receptor modulators towards clinical development in 2026, with best-in-class potential across a range of prevalent and underserved neuropsychiatric disorders.
The founding team of Prof. John Atack (Chief Translational Officer), Prof. Simon Ward (Chief Scientific Officer) and Ruth McKernan CBE (Executive Chair) has extensive experience in company creation and central nervous system drug development. Draig is now an established international company whose executive team brings deep biotech and pharma experience, and includes Inder Kaul MD (Chief Medical Officer, formerly at Bristol Myers Squibb and Karuna Therapeutics), Florian Islinger MD (Chief Commercial Strategy Officer, formerly at Roche) and David Watson (Chief Operating Officer, formerly at Biogen).
More details on members of the Board of Directors and the Scientific Advisory Board can be found on the Draig website at www.draigtherapeutics.com
ENDS
For further information:
Mark Swallow, Sandi Greenwood
E-mail: draigtx@medistrava.com
Draig Therapeutics
E-mail: rmckernan@draigtherapeutics.com
About Draig Therapeutics
Draig Therapeutics is a clinical-stage company with a mission to transform treatments in Neuropsychiatry. The company is leveraging its founders’ unique scientific expertise in modulating the core glutamate / GABA pathways that play a critical role in neuropsychiatric diseases to advance a pipeline of groundbreaking therapies designed to address large unmet patient needs, including in Major Depressive Disorder (MDD).
Draig is the Welsh word for ‘dragon’ and it reflects the company’s origins in Wales. The name and logo were inspired by this heritage, reflecting its scientific roots stemming from Cardiff University.
Draig was co-founded by Cardiff University and SV Health Investors, which led the seed financing with ICG, and is backed by other leading healthcare venture firms including Access Biotechnology, Canaan Partners, SR One, Sanofi Ventures and Schroders Capital. For more information, please visit www.draigtherapeutics.com
About Access Biotechnology
Access Biotechnology is the life science investment arm of Access Industries, and invests in transformative therapies for conditions with high unmet needs and that have the potential to meaningfully impact human health. www.accessindustries.com/biotechnology
About Access Industries
Access Industries is a privately held global investment firm founded in 1986 by businessman and philanthropist Len Blavatnik. Access identifies companies with strong business models and management teams, and optimizes their value through financial backing and expertise. Access currently manages more than $35 billion in perpetual, flexible capital, guided by a team of investment professionals. The portfolio includes substantial investments across biotechnology, entertainment, global media, real estate, technology, and other sectors. www.accessindustries.com
About SV Health Investors
SV Health Investors is a leading healthcare fund manager committed to investing in tomorrow’s healthcare breakthroughs. The SV funds invest across stages, geographic regions, and sectors, with expertise spanning biotechnology, dementia, medical devices, healthcare growth and healthcare technology. With approximately $2bn in assets under management and a truly transatlantic presence with offices in London and Boston, SV has built an extensive network of talented investment professionals and experienced industry veterans. Since its founding in 1993, SV has invested in, created and built more than 200 companies attracting global talent, entrepreneurs and pharma partners. To date, these investments have resulted in the licensing of 27 novel drugs and
six new drug classes able to treat indications with unmet medical needs and deliver positive impact to patients. For more information, please visit www.svhealthinvestors.com.
About ICG
ICG (LSE: ICG) is a global alternative asset manager with $112bn* in AUM and more than three decades of experience generating attractive returns. We operate from over 20 locations globally and invest our clients' capital across Structured Capital; Private Equity Secondaries; Private Debt; Credit; and Real Assets. Our exceptional people originate differentiated opportunities, invest responsibly, and deliver long-term value. We partner with management teams, founders, and business owners in a creative and solutions-focused approach, supporting them with our expertise and flexible capital. For more information visit our website and follow us on LinkedIn. *As at 31 March 2025.
For more information about our other investors, please see the following websites:

SAN FRANCISCO – June 5, 2025 – Omada Health, the virtual between-visit healthcare provider, today announced the pricing of its initial public offering of 7,900,000 shares of its common stock, at a public offering price of $19.00 per share. In addition, Omada Health has granted the underwriters a 30-day option to purchase up to an additional 1,185,000 shares of common stock at the initial public offering price, less underwriting discounts and commissions. The shares are expected to begin trading on the Nasdaq Global Select Market on June 6, 2025, under the ticker symbol “OMDA.” The offering is expected to close on June 9, 2025, subject to customary closing conditions.
Morgan Stanley, Goldman Sachs & Co. LLC and J.P. Morgan are acting as lead book-running managers for the proposed offering. Barclays and Evercore ISI are acting as joint book-running managers for the proposed offering. Canaccord Genuity and Needham & Company are acting as co-managers for the proposed offering.
A registration statement relating to these securities has been filed with and declared effective by the Securities and Exchange Commission. This offering is being made only by means of a prospectus, copies of which may be obtained, when available, from: Morgan Stanley & Co. LLC, Attention: Prospectus Department, 180 Varick Street, 2nd Floor, New York, New York 10014, or by email at prospectus@morganstanley.com; Goldman Sachs & Co. LLC, Attention: Prospectus Department, 200 West Street, New York, New York 10282, by telephone at (866) 471-2526, by facsimile at (212) 902-9316, or by email at prospectus-ny@ny.email.gs.com; or J.P. Morgan Securities LLC, c/o Broadridge Financial Solutions, 1155 Long Island Avenue, Edgewood, New York 11717, or by email at prospectus-eq_fi@jpmorgan.com and postsalemanualrequests@broadridge.com.
This press release shall not constitute an offer to sell or the solicitation of an offer to buy these securities, nor shall there be any sale of these securities in any state or jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction.
About Omada Health
Omada Health is a virtual-first healthcare provider that nurtures lifelong health, one day at a time. Omada care teams implement clinically-validated behavior change protocols for individuals living with diabetes, hypertension, prediabetes, and musculoskeletal issues. With more than a decade of experience and data, and 29 peer-reviewed publications that showcase its clinical and economic results, Omada is designed to help improve health outcomes and contain healthcare costs. Omada’s scope exceeds 2,000 customers, including health plans, health systems, and employers ranging in size from small businesses to Fortune 500s.
Contacts
Rose Ramseth
Allan Kells
- Pioneering small-molecule program, NT-0796, targets chronic inflammation and its fundamental role in the complex cycle of weight regulation
- Oral, brain-penetrant NLRP3 inflammasome inhibitor being investigated as monotherapy in 24-week Phase 2 RESOLVE-1 trial in 160 patients with obesity, with and without type 2 diabetes
- Combination trial planned with GLP-1RA to improve efficacy and tolerability of marketed anti-obesity treatments
- NT-0796 uniquely positioned as an oral, well-tolerated, small molecule therapeutic to enable sustained and healthy weight loss
- Acceleration of NT-0796 program follows $50 million Series D financing
Philadelphia, PA, June 4, 2025 - NodThera, a leading clinical-stage biotech delivering a paradigm shift in the treatment of chronic inflammatory diseases through selective modulation of the NLRP3 inflammasome, today announces that the first patients have been dosed in its Phase 2 RESOLVE-1 (RESolution Of infLammation to treat obesity and cardioVascular disEase) clinical trial investigating the potential of its lead candidate, oral NLRP3 inflammasome inhibitor NT-0796, in patients with obesity.
NodThera’s pioneering approach to obesity treatment is built on the growing understanding of the impact of chronic inflammation on the body and its fundamental role in the complex cycle of weight regulation which balances calorie intake, energy expenditure and satiety. While current drugs have made significant achievements in the management of obesity, their mechanisms primarily suppress the desire to eat (calorie restriction) or they promote a feeling of fullness, ultimately reducing food intake. NodThera’s approach goes beyond the reduction of food intake by resetting the body’s weight regulatory system and correcting the underlying imbalance, to return the body to its natural metabolic state.
Daniel Swisher, Chief Executive Officer of NodThera, commented: “With its novel molecular mechanism, the convenience of oral dosing without titration, and a well-tolerated safety profile, this fully brain-penetrant NLRP3 inflammasome inhibitor is projected to have a transformative impact on obesity by bringing the body back to balance. I am delighted that, following our recently closed $50 million Series D financing, we have accelerated our lead program into this robust Phase 2 trial, building on an impressive preclinical and clinical data set. This will support the progression of NT-0796 as an enhanced treatment for weight management, to enable patients to achieve sustained and healthy weight loss.”
RESOLVE-1 is a randomized, double-blind, placebo-controlled Phase 2 trial that will evaluate the efficacy and safety of NT-0796 in patients with obesity, with or without type 2 diabetes, over 24 weeks. 160 patients are expected to be randomized into three treatment groups, receiving twice daily oral NT-0796, once daily oral NT-0796 or placebo. The trial’s primary endpoint is the change in body weight in participants from baseline to week 24. Secondary endpoints include the impact of NT-0796 on body composition, metabolic biomarkers, and HbA1c levels in participants with type 2 diabetes. Headline data from the study are anticipated in Q2 2026.
Returning the body to its natural metabolic balance – homeostasis
Earlier preclinical and clinical work undertaken by the Company has demonstrated that NLRP3, an intracellular sensor and master switch that modulates inflammation, plays a key role in controlling obesity and obesity-associated inflammation. The fully brain-penetrant NT-0796 reduces inflammation in the brain, as well as throughout the body, to restore multiple dysregulated cardiometabolic pathways. The inhibition of NLRP3 results in the loss of fat mass, while preserving lean muscle mass, and restores the body’s natural metabolic balance (homeostasis), enabling sustainable, enhanced and healthy weight loss.
Positive clinical data announced by the Company in June 2024, from a Phase 1b/2a cardiovascular risk study in patients with obesity, confirmed NT-0796’s profound anti-inflammatory effect and supported its anti-obesity potential. This followed the publication of preclinical data in the Journal of Pharmacology and Experimental Therapeutics, demonstrating the potential of NT-0796 to reverse diet-induced obesity and inflammation in an animal model of the disease, matching weight loss driven by the GLP-1 receptor agonist, semaglutide. Most recently, data published in the journal Obesity showed the enhanced weight loss effect of NT-0796 when combined with semaglutide in a preclinical obesity model. The potential of NT-0796 to improve the efficacy and tolerability of existing GLP-1RA obesity therapies will be further investigated in NodThera’s upcoming Phase 2 combination trial, RESOLVE-2, expected to commence in H2 2025.
Alan Watt, President and Chief Scientific Officer of NodThera, added: “Through central NLRP3 inhibition and the correction of multiple dysregulated pathways in the brain and throughout the body, NT-0796 has the potential to deliver robust weight loss and broad cardiometabolic benefits that go beyond those seen with current therapies. NodThera’s treatment approach, restoring the body’s natural metabolic balance, would be game-changing.”
For more information about NodThera please contact:
NodThera
Tel: +44 (0) 1223 608130
Email: info@nodthera.com
ICR Healthcare
Amber Fennell, David Daley
Tel: +44 (0)20 3709 5700
Email: nodthera@icrhealthcare.com
About NodThera
NodThera is a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases. Led by an experienced management team, NodThera is combining a deep understanding of NLRP3 inhibition, pharmaceutical neuroscience expertise and precision chemistry. Its two lead clinical candidates are oral, small molecule NLRP3 inflammasome inhibitors, which have demonstrated differentiated, potentially best-in-class clinical profiles with significant anti-inflammatory effects and high brain penetration, offering distinct opportunities to treat multiple indications. The Company is backed by top-tier investors including 5AM Ventures, Blue Owl Capital, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Philadelphia, Pennsylvania, with additional operations in Cambridge, UK. Learn more at www.nodthera.com or follow the Company on LinkedIn.
- Financing co-led by new investors EQT Life Sciences and Sanofi Ventures, with participation from Roche Venture Fund, as well as all existing investors
- Proceeds will support clinical development of lead program SB-007 in Stargardt disease
- Funding will also advance a broader pipeline of genetic medicines targeting indications in ophthalmology, neurology, and other undisclosed therapeutic areas
BARCELONA, SPAIN, 11 June 2025 – SpliceBio, a clinical-stage genetic medicines company pioneering Protein Splicing to address diseases caused by mutations in large genes, today announced the close of a $135 million Series B financing co-led by new investors EQT Life Sciences and Sanofi Ventures, with participation from Roche Venture Fund, as well as all existing investors: New Enterprise Associates, UCB Ventures, Ysios Capital, Gilde Healthcare, Novartis Venture Fund, and Asabys Partners.
The funding will be used to advance the clinical development of SpliceBio’s lead gene therapy candidate, SB-007 for Stargardt disease, including the ongoing interventional Phase 1/2 ASTRA study and the observational POLARIS study. SB-007 is the first dual adeno-associated viral (AAV) gene therapy cleared by the Food and Drug Administration (FDA) to enter clinical development for Stargardt disease. SB-007 has also received regulatory clearance for clinical development from the UK Medicines and Healthcare products Regulatory Agency (MHRA).
Stargardt disease is an inherited retinal disorder caused by mutations in the ABCA4 gene that leads to progressive vision loss and blindness, with no approved treatments available. SB-007 is designed to address the underlying genetic cause of the disease by producing a functional copy of the full-length ABCA4 protein with the potential to treat all patients, regardless of their specific ABCA4 mutation. The proceeds will also be used to accelerate SpliceBio’s pipeline of AAV gene therapy programs in ophthalmology, neurology, and other undisclosed indications that utilise the company’s proprietary Protein Splicing platform.
“This financing marks a pivotal milestone for SpliceBio as we advance the clinical development of SB-007 for Stargardt disease and continue to expand our pipeline across ophthalmology, neurology and beyond,” said Miquel Vila-Perelló, Ph.D., Chief Executive Officer and Co-Founder of SpliceBio. “The support from such high-quality investors underscores the strength of our programs and our unique Protein Splicing platform and its potential to unlock gene therapies for diseases that remain untreatable today. We are building a company positioned to lead the next wave of genetic medicines.”
SpliceBio is redefining and expanding the scope of diseases that can be tackled with gene therapies by addressing a fundamental limitation of AAV vectors in their inability to deliver genes that exceed their limited packaging capacity of 4.7 kilobases. Many genetic disorders remain untreatable because the necessary gene is too large to fit into the AAV vectors. SpliceBio’s unique Protein Splicing platform leverages the use of a family of proprietary, engineered proteins called inteins, originally developed at Princeton University. The company’s technology enables the splitting of the gene into two (or more) transgenes that are then delivered using dual AAV vectors. Once inside the cell, the DNA of each transgene is transcribed into messenger RNA and translated into protein. SpliceBio’s engineered inteins are designed to then assemble the full-length protein that is needed to treat the disease.
Daniela Begolo, Managing Director at EQT Life Sciences, commented: “We are proud to support SpliceBio, a pioneer among the next-generation of genetic medicine companies. Its Protein Splicing platform is designed to offer a novel solution to deliver large genes with AAV, one of the field's most pressing challenges, and exemplifies our commitment to backing transformational science that can meaningfully benefit patients' lives.”
Laia Crespo, Partner at Sanofi Ventures, remarked: “With compelling data for its lead program, SB-007, and a highly differentiated platform, we are excited to support SpliceBio as it tackles a fundamental challenge for genetic medicines. By enabling the delivery of large and complex genes through its novel AAV vector Protein Splicing technology, SpliceBio has the potential to make a significant impact on the field of gene therapy and to deliver best-in-class therapies to patients.”
Carole Nuechterlein, Head of Roche Venture Fund, added: “We are impressed by the team’s strong execution, the momentum behind SB-007 in Stargardt disease, and the platform’s potential to unlock a new class of genetic medicines. We are proud to support SpliceBio at this pivotal stage of growth as they advance their lead program through clinical development and explore additional high-impact indications.”
In connection with the financing, Daniela Begolo, Managing Director at EQT Life Sciences, Laia Crespo, Partner at Sanofi Ventures, and Carole Nuechterlein, Head of Roche Venture Fund, will join the SpliceBio Board of Directors.
-Ends-
For further information:
Optimum Strategic Communications
Mary Clark, Zoe Bolt, Elena Bates
Email: splicebio@optimumcomms.com
Tel: +44 (0) 20 3882 9621
About SpliceBio
SpliceBio is a clinical-stage genetic medicines company pioneering Protein Splicing to address diseases caused by mutations in large genes. The Company’s lead program, SB-007, targets the root cause of Stargardt disease, a genetic eye disease that causes blindness in children and adults. SpliceBio’s pipeline comprises additional gene therapy programs across therapeutic areas, including ophthalmology and neurology. SpliceBio’s platform is based on technology developed in the Muir Lab at Princeton University after more than 20 years of pioneering intein, Protein Splicing, and protein engineering research. For additional information, please visit www.splice.bio.
About SB-007
SB-007 is an investigational Protein Splicing dual AAV gene therapy in development for the treatment of Stargardt disease. It is designed to restore expression of the native full-length ABCA4 protein in the retina. SB-007 has been granted Orphan Drug Designation from both the FDA in the US and the European Commission in Europe. In December 2024, SB-007 received FDA IND clearance, marking the first-ever clearance for a dual AAV gene therapy in Stargardt disease. Alongside initiation of the Phase 1/2 ASTRA study, with the announcement of the first patient dosed in March 2025, SpliceBio continues to advance POLARIS, a natural history study of the disease. Both studies are actively recruiting. For more information or to enquire about participation in the studies, please visit https://splice.bio/clinical/.
About EQT Life Sciences
EQT Life Sciences was formed in 2022 following an integration of LSP, a leading European life sciences and healthcare venture capital firm, into the EQT platform. As LSP, the firm raised over EUR 3.0 billion (USD 3.5 billion) and supported the growth of more than 150 companies since it started to invest over 30 years ago. With a dedicated team of highly experienced investment professionals, coming from backgrounds in medicine, science, business, and finance, EQT Life Sciences backs the smartest inventors who have ideas that could truly make a difference for patients. More info: www.eqtgroup.com. Follow EQT on LinkedIn, Twitter, YouTube and Instagram.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi, focused on investing in promising early-stage healthcare companies. The firm supports pioneering innovations in biotechnology, digital health, and life sciences aligning with Sanofi’s mission to bring life-changing treatments to patients worldwide. For more information visit: www.sanofiventures.com
About Roche Venture Fund
The Roche Venture Fund is the corporate venture fund of Roche and invests in innovative life science companies. Over the past 20 years, the Roche Venture Fund has invested in over 60 companies globally and currently has a portfolio of around 30 companies located in 10 countries. As part of a multinational healthcare company, the Roche Venture Fund has access to considerable expertise both internally and externally and co-invests with leading venture funds, including other corporate venture funds, on a regular basis.
https://www.roche.com/venturefund

Funding is part of the call for proposals “Innovations in Biotherapies and Bioproduction”, coordinated by the Health Innovation Agency under France 2030 strategy, operated on behalf of the French government by Bpifrance.
Paris, France – [April 14, 2025] – Eligo Bioscience, a biotechnology company pioneering the development of novel genetic medicines, today announced it has secured a $5 million grant from the French government to accelerate the advancement and scaling of Eligo’s proprietary topical gene delivery platform, designed to enable local production of therapeutic biologics directly by skin-resident bacteria.
This grant follows recent key milestones for Eligo, including the publication of its microbiome baseediting platform in Nature, securing multiple foundational patents covering in situ editing of the skin microbiome, and successfully expanding its Series B funding round to $35 million. This latest nondilutive financial support strengthens Eligo’s capacity to rapidly advance its clinical pipeline.
Scaling up and advancing towards the clinic
Building on the development of its first-generation CRISPR-based topical therapeutic targeting moderate to severe acne vulgaris, Eligo will now optimize and scale up the bioproduction process of its innovative gene delivery vector ahead of later-stage clinical trials. In this endeavor, Eligo is partnering with Biose Industries, a globally recognized CDMO specializing in microbial fermentation. Biose’s extensive GMP expertise in scaling complex microbial therapeutics will be instrumental in accelerating Eligo’s progress toward clinical validation.
“This funding empowers us to establish a robust bioproduction process for our first-in-class topical microbiome-targeting gene-editing therapies, laying the groundwork for expanding into a wide range of immuno-dermatological indications,” said Dr. Xavier Duportet, CEO and Co-Founder of Eligo Bioscience. “It reinforces our ambition to deliver truly transformative therapeutic solutions, addressing critical medical challenges faced by millions of patients worldwide.”
Expansion to immuno-dermatology targets
The skin microbiome plays a pivotal role in cutaneous immune regulation, and commensal bacteria residing within the hair follicle lie in immediate proximity to resident immune cells.
Eligo’s unique in-situ delivery modality enables to perform in-situ genetic engineering of these commensal bacteria, turning them as localized bioreactors that can express high-potency biologics precisely where immune dysregulation occurs.
The new funding will support the exploration of multiple therapeutic payloads and the build-out of a diversified immuno-dermatology pipeline targeting chronic inflammatory and immune-mediated skin diseases with significant unmet need.
For more information about Eligo Bioscience, France 2030, and Bpifrance: eligo.bio/france2030
Collaboration with UMass Chan Medical School confirmed FMR1-217 as well as an RNA-targeted mechanism to restore functional FMRP protein to develop potential treatments for FXS
QurAlis’ preliminary data suggest feasibility of biomarker to detect mis-splicing of FMR1 in FXS; company advancing FMR1-217 as precision medicine target in up to 80 percent of FXS patients
Company applying its FlexASO® platform and expertise of splicing targets toward having a candidate nominated for IND-enabling studies in the near future
CAMBRIDGE, Mass., May 15, 2025 – QurAlis Corporation (“QurAlis”), a clinical-stage biotechnology company driving scientific breakthroughs into powerful precision medicines that have the potential to alter the trajectory of neurodegenerative and neurological diseases, today announced it has entered into an exclusive license agreement with UMass Chan Medical School (“UMass Chan”) on a novel RNA-targeted mechanism confirmed to restore functional protein for Fragile X syndrome (FXS).
Fragile X syndrome is the leading inherited form of intellectual disability and the most common single genetic cause of autism. It is a genetic condition caused by a mutation of a single gene – Fragile X messenger ribonucleoprotein 1 (FMR1) – on the X chromosome. This mutation of FMR1 causes a range of developmental problems including learning disabilities, behavioral challenges, and cognitive impairment.
QurAlis’ exclusive license agreement is a result of its 2024 partnership and collaboration with UMass Chan to explore the biology of FXS to determine and confirm relevant targets that could enable antisense oligonucleotide (ASO)-mediated correction for FXS. QurAlis leveraged its deep understanding, knowledge and expertise in developing ASOs as part of the collaboration. QurAlis confirmed the findings from the original publication of the UMass Chan researchers and is advancing FMR1 as a precision medicine target in up to 80 percent of FXS patients. The mis-spliced form of FMR1, designated as FMR1-217, is widely expressed throughout cortical brain areas affected in FXS and can be measured in blood and cerebrospinal fluid. Preliminary data suggest biomarker feasibility to detect mis-splicing of FMR1 in patients with FXS.
“FXS is a devastating neurodevelopmental disorder with no effective disease-modifying therapies available. Our initial partnership with UMass Chan confirmed that FMR1-217 is a validated genetic target for FXS,” said Kasper Roet, Ph.D., chief executive officer and co-founder of QurAlis. “This groundbreaking discovery of a novel RNA-targeted mechanism to restore functional protein for FXS and the feasibility of a biomarker to detect mis-splicing of FMR1 in FXS patients opens up a completely new type of therapeutic approach through splice correction. We look forward to applying QurAlis’ FlexASO® platform and deep knowledge and expertise of ASO splicing targets toward having a candidate nominated for IND-enabling studies in the near future, so that we can bring a potential new precision medicine option to patients.”
Joel Richter, Ph.D., the Arthur F. Koskinas Chair in Neuroscience and professor of molecular medicine at UMass Chan, and colleagues Sneha Shah, Ph.D., and Jonathan K. Watts, Ph.D., together with Elizabeth Berry-Kravis, M.D., Ph.D., at Rush University Medical Center, have shown that aberrant alternative splicing, or mis-splicing, of messenger RNA (mRNA) plays a fundamental role in FXS. In a seminal publication by the group, it was revealed that in FXS patients, FMR1 mRNA is still being expressed, but is mis-spliced, comprising a short, truncated alternative mRNA variant called FMR1-217 which results in nonfunctional FMRP protein expression. Working with patient-derived cells, Dr. Richter’s lab and Dr. BerryKravis initially demonstrated that ASOs can successfully inhibit the mis-splicing, reduce expression of FMR1-217, rescue proper FMR1 mRNA, and restore FMRP protein expression.
“This is a meaningful step in the process of taking basic biological discoveries and turning them into practical therapies that can benefit patients in the clinic,” said Dr. Richter. “QurAlis’ platform and expertise in neurodegenerative disorders are industry leading and well positioned to address the mis-splicing of FMR1 RNA and restore functional FMRP protein expression. This partnership has not only validated our years-long research but also has resulted in the confirmation of a novel target for FXS, which we hope will lead to much-needed treatment options for FXS patients and their families.”
Dr. Berry-Kravis added, “I am very excited that we will be able to continue development of this potential genetically based disease-modifying FMRP-restoring therapy that is expected to have a major impact on the FXS field and the spectrum of treatment options available to improve function in people with FXS.”
An orphan disease, FXS affects approximately 87,000 individuals in the U.S. alone – one in 4,000 men and one in 6,000 women. Though FXS occurs in both genders, males are more frequently affected than females, and generally with greater severity. In addition to intellectual disability, FXS patients endure a wide range of disabling symptoms including severe anxiety, social aversion, hyperactivity and attention deficit, sensory hypersensitivity, aggression, developmental seizures, and others. There are no effective disease-modifying therapies currently available for FXS.
ASOs are short, engineered single-stranded DNA/RNA molecules that can selectively bind RNA to regulate its expression in the cell. ASO technology has been leading in the field of protein regulation and has since allowed us to develop treatments for neurodegenerative disease by changing the expression of genes connected to the disease.
QurAlis’ FlexASO® platform was developed to generate splice-switching ASOs with improved potency, increased therapeutic index and improved bio-distribution. This bespoke platform has the potential to tackle the spectrum of neurodegenerative and neurological diseases.
About QurAlis Corporation
At QurAlis, we are neuro pioneers on a quest to cure, boldly seeking to translate scientific breakthroughs into powerful precision medicines. We work collaboratively with a relentless pursuit of knowledge, precise attention to craft, and compassion to discover and develop medicines that have the potential to transform the lives of people living with neurodegenerative and neurological diseases. QurAlis is the leader in development of precision therapies for amyotrophic lateral sclerosis (ALS). In addition to ALS, QurAlis is advancing a robust precision medicine pipeline to bring effective disease-modifying therapeutics to patients suffering from severe diseases defined by genetics and clinical biomarkers. For more information, please visit www.quralis.com or follow us on X @QurAlisCo or LinkedIn.
Contact:
Kathy Vincent
kathy.vincent@quralis.com
310-403-8951
CAMBRIDGE, Mass. – May 15, 2025 – Quiver Bioscience (“Quiver”) and QurAlis Corporation (“QurAlis”), today announced that the companies have entered into a research collaboration to advance a novel gene-targeted therapeutic approach for the treatment of Fragile X syndrome (FXS). The goal of the collaboration is to combine Quiver’s unique “Genomic Positioning System” (GPS) drug discovery platform with QurAlis’ expertise in developing next-generation precision medicines for neurodegenerative and neurological diseases to build a foundational data package in support of advancing a potentially transformative therapeutic for FXS.
Quiver’s GPS platform integrates unique-in-world, scalable, human neuronal electrophysiology data (the ‘language’ of the brain) with artificial intelligence and machine learning (AI/ML) to drive novel insights into disease biology and enable optimized drug discovery. Quiver has successfully applied its GPS approach to a variety of central nervous system (CNS) disorders and recently published modeling and drug discovery efforts in FXS.
“Our platform technology is uniquely suited to improving understanding of the molecular and cellular basis of neurogenetic disorders such as FXS. We are excited to embark on this partnership with QurAlis which aspires to bring about groundbreaking therapies for the FXS community,” said Graham Dempsey, Ph.D., co-founder and CEO of Quiver Bioscience.
“FXS is a devastating neurodevelopmental disease. It is the leading inherited form of intellectual disability and known cause of autism for which there are no disease-modifying therapies,” said Kasper Roet, Ph.D., CEO and co-founder of QurAlis. “We look forward to this research collaboration with Quiver. The combination of enabling technologies and drug development experience built through this partnership holds great promise for progressing novel therapeutics for FXS, for which there exists a significant unmet medical need.”
FXS, the leading genetic form of intellectual disability and autism spectrum disorder, is caused by loss of the FMR1 encoded protein Fragile X Messenger Ribonucleoprotein (FMRP). It currently affects approximately 87,000 individuals in the U.S. alone – occurring at an incidence of 1 in 4,000 males and 1 in 6,000 females. In addition to intellectual disability, FXS symptoms include delays in development, seizures, speech difficulties, hyperactivity and ahention deficit, severe anxiety, and others. There are no disease-modifying therapies currently available for FXS.
Destum Partners acted as transaction advisor to Quiver Bioscience.
About Quiver Bioscience
Quiver Bioscience is a technology-driven company established to create transformational medicines for the brain while simultaneously uncovering new biology and novel, effective drug targets. Using advanced single-cell imaging and multi-omics, we are building the world's most information-rich neuronal insight map via our "Genomic Positioning System." Our approach integrates cukng-edge scalable human models, state-of-the-art technology and proprietary engineering, and learning and surrogate AI/ML models to identify novel therapeutic targets and the best candidate molecules to deliver new and meaningful therapeutics to patients. For information, including additional publications describing application of Quiver’s GPS to drug discovery, visit www.quiverbioscience.com or follow us on LinkedIn.
About QurAlis Corporation
At QurAlis, we are neuro pioneers on a quest to cure, boldly seeking to translate scientific breakthroughs into powerful precision medicines. We work collaboratively with a relentless pursuit of knowledge, precise ahention to cral, and compassion to discover and develop medicines that have the potential to transform the lives of people living with neurodegenerative and neurological diseases. QurAlis is the leader in development of precision therapies for amyotrophic lateral sclerosis (ALS). In addition to ALS, QurAlis is advancing a robust precision medicine pipeline to bring effective disease-modifying therapeutics to patients suffering from severe diseases defined by genetics and clinical biomarkers. For more information, please visit www.quralis.com or follow us on X @QurAlisCo or LinkedIn.
Media Contact:
For Quiver Bioscience:
Noélle Germain
noelle.germain@quiverbioscience.com
For QurAlis Corporation:
Kathy Vincent
kathy.vincent@quralis.com

SAN RAFAEL, Calif. and BOSTON, May 16, 2025 /PRNewswire/ -- BioMarin Pharmaceutical Inc. (Nasdaq: BMRN) and Inozyme Pharma, Inc. (Nasdaq: INZY) announced today that BioMarin has entered into a definitive agreement to acquire Inozyme for $4.00 per share in an all-cash transaction for a total consideration of approximately $270 million. The transaction has been unanimously approved by the Boards of Directors of both companies and is expected to close in the third quarter of 2025, subject to regulatory approval, successful completion of a tender offer and other customary closing conditions.
The acquisition will strengthen BioMarin's enzyme therapies portfolio, adding a late-stage enzyme replacement therapy, INZ-701, which is currently being assessed for the treatment of ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1) Deficiency, a rare, serious and progressive genetic condition that affects blood vessels, soft tissues and bones. The condition is associated with increased cardiovascular mortality risk across all age groups, especially in infants. It is also associated with severe rickets and osteomalacia in children and adults. Data from the first Phase 3 pivotal study of INZ-701 in children is expected in early 2026, with potential regulatory approval in 2027.
"BioMarin has been deeply committed to advancing enzyme therapies for children and adults living with serious genetic conditions for more than 25 years, and today's agreement builds on our legacy," said Alexander Hardy, President and Chief Executive Officer of BioMarin. "This acquisition brings to BioMarin an important medicine that has the potential to be the first treatment for children and adults with ENPP1 Deficiency, improving care for people living with this serious condition. As BioMarin continues our transformation and delivers on our corporate strategy, we will continue to evaluate external innovation alongside internal innovation. We are in a strong financial position to bring in additional assets as we accelerate the development of medicines for patients with significant unmet need."
"Today's announcement gives greater hope to patients who may benefit from INZ-701, a potentially transformative therapy that aims to address the underlying causes and systemic impacts of ENPP1 Deficiency," said Douglas A. Treco, Ph.D., Chief Executive Officer and Chairman
of Inozyme. "BioMarin has paved the way over the past two and a half decades, successfully launching five first-in-disease enzyme therapies. I'd like to thank the team at Inozyme and our partners for their outstanding work and dedication, as we pass this important potentially life-changing therapy to the leading innovator in genetically defined conditions."
INZ-701 and ENPP1 Deficiency
INZ-701 is a potential first-in-class, subcutaneous enzyme replacement therapy that is being developed for infants, pediatric and adult patients with ENPP1 Deficiency across a continuum of phenotypes. In addition to the ongoing Phase 3 pivotal study in children, Inozyme is currently enrolling infants in a pivotal study, and a supportive study for adolescents and adults is being planned.
In a Phase 1/2 study of adults living with ENPP1 Deficiency, INZ-701 demonstrated a favorable safety profile, with no serious adverse events attributed to INZ-701. Improvements in pyrophosphate levels, bone mineralization biomarkers and quality of life were all observed, suggesting prospect for benefit in patients.
ENPP1 Deficiency is a lifelong, rare, progressive, multisystemic condition, caused by mutations in the ENPP1 gene, leading to loss of ENPP1 enzymatic activity that results in low pyrophosphate, upregulation of fibroblast growth factor-23 and phosphate wasting. The condition affects blood vessels, soft tissues and bones. It is associated with high risk of cardiovascular mortality in patients of all ages, especially infants. It is also associated with severe rickets and osteomalacia in children and adults. Patients require considerable multidisciplinary lifelong medical and surgical management of complications. Currently there are no approved therapies for ENPP1 Deficiency.
Terms of the Transaction
Under the terms of the merger agreement, BioMarin will promptly commence a cash tender offer to acquire all of the outstanding shares of Inozyme common stock at a price of $4.00 per share. Inozyme's Board of Directors unanimously recommends that Inozyme's stockholders tender their shares in the tender offer.
The consummation of the tender offer is subject to customary closing conditions, including the tender of at least a majority of the outstanding shares of Inozyme, the expiration or termination of the waiting period under the Hart-Scott-Rodino Antitrust Improvements Act of 1976, and other customary conditions. Following the successful completion of the tender offer, a wholly owned subsidiary of BioMarin will merge with Inozyme and the outstanding Inozyme shares not tendered in the tender offer will be converted into the right to receive the same $4.00 per share in cash paid in the tender offer.
The transaction is not subject to any financing condition.
BioMarin also today reaffirmed previously provided full-year 2025 financial guidance, excluding the impact of the accounting treatment of this transaction in accordance with Generally Accepted Accounting Principles (GAAP) upon closing, as well as its plan to achieve 40% Non-GAAP Operating Margin in 2026.
Advisors
Goldman Sachs & Co. LLC is acting as exclusive financial advisor to BioMarin, and Cooley LLP is serving as legal counsel. Centerview Partners LLC is acting as exclusive financial advisor to Inozyme, and Goodwin Procter LLP is serving as legal counsel.
Conference Call
BioMarin will host a conference call and webcast to discuss the acquisition today, May 16, 2025, at 8:45 a.m. ET. This event can be accessed through this link or on the investor section of the BioMarin website at www.biomarin.com. U.S./Canada Dial-in Number: 888-596-4144 Replay Dial-in Number: 800-770-2030 International Dial-in Number: 646-968-2525 Replay International Dial-in Number: 609-800-9909 Conference ID: 2239224.
About BioMarin
BioMarin is a global biotechnology company dedicated to translating the promise of genetic discovery into medicines that make a profound impact on the life of each patient. The San Rafael, California-based company, founded in 1997, has a proven track record of innovation with eight commercial therapies and a strong clinical and preclinical pipeline. Using a distinctive approach to drug discovery and development, BioMarin seeks to unleash the full potential of genetic science by pursuing category-defining medicines that offer new possibilities for people living with genetically defined conditions around the world.
To learn more, please visit www.biomarin.com.
About Inozyme
Inozyme Pharma is a clinical-stage biopharmaceutical company, with approximately 50 employees based in Boston. The company is dedicated to developing innovative therapeutics that target the PPi-Adenosine Pathway, a key regulator of bone health and blood vessel function. Disruptions in this pathway underlie a range of severe diseases, including ENPP1 Deficiency. Our lead investigational therapy, INZ-701, is an ENPP1 Fc fusion protein enzyme replacement therapy designed to restore pyrophosphate and adenosine levels. INZ-701 is currently in late-stage clinical development in ENPP1 Deficiency, with the potential to expand into additional indications where deficiencies in the Pyrophosphate-Adenosine Pathway contribute to disease pathology, including ABCC6 Deficiency and calciphylaxis. Through our pioneering work, we aim to transform treatment options for patients affected by these devastating conditions.
To learn more, please visit www.inozyme.com.
Forward-Looking Non-GAAP Financial Information
As described above, today BioMarin reaffirmed its plan to achieve 40% Non-GAAP Operating Margin in 2026. BioMarin does not provide guidance for GAAP reported financial measures (other than revenue) or a reconciliation of forward-looking Non-GAAP financial measures to the most directly comparable GAAP reported financial measures because BioMarin is unable to predict with reasonable certainty the financial impact of changes resulting from its strategic portfolio and business operating model reviews; potential future asset impairments; gains and losses on investments; and other unusual gains and losses without unreasonable effort. These items are uncertain, depend on various factors, and could have a material impact on GAAP reported results for the guidance period. As such, any reconciliations provided would imply a degree of precision that could be confusing or misleading to investors.
Non-GAAP Information
Non-GAAP Operating Margin percentage is defined by BioMarin as GAAP Income from Operations, excluding amortization of intangible assets, stock-based compensation expense and, in certain periods, certain other specified items, divided by GAAP Total Revenues.
BioMarin regularly uses both GAAP and Non-GAAP results and expectations internally to assess its financial operating performance and evaluate key business decisions related to its principal business activities: the discovery, development, manufacturing, marketing and sale of innovative biologic therapies. Because Non-GAAP Operating Margin percentage is an important internal measurement for BioMarin, BioMarin believes that providing this information in conjunction with BioMarin's GAAP information enhances investors' and analysts' ability to meaningfully compare BioMarin's results from period to period and to its forward-looking guidance, and to identify operating trends in BioMarin's principal business.
Non-GAAP financial measures are not meant to be considered in isolation or as a substitute for, or superior to comparable GAAP measures and should be read in conjunction with the consolidated financial information prepared in accordance with GAAP. Investors should note that the Non-GAAP information is not prepared under any comprehensive set of accounting rules or principles and does not reflect all of the amounts associated with BioMarin's results of operations as determined in accordance with GAAP. Investors should also note that these Non-GAAP financial measures have no standardized meaning prescribed by GAAP and, therefore, have limits in their usefulness to investors. In addition, from time to time in the future there may be other items that BioMarin may exclude for purposes of its Non-GAAP financial measures; likewise, BioMarin may in the future cease to exclude items that it has historically excluded for purposes of its Non-GAAP financial measures. Because of the non-standardized definitions, the Non-GAAP financial measure as used by BioMarin in this press release may be calculated differently from, and therefore may not be directly comparable to, similarly titled measures used by other companies.
Forward-Looking Statements
This press release and the associated conference call contain forward-looking statements about, among other things, the proposed acquisition of Inozyme Pharma, Inc. (Inozyme) by BioMarin Pharmaceutical Inc. (BioMarin) and the business prospects of Inozyme and BioMarin, including, without limitation, statements about: the anticipated occurrence, manner and timing of the proposed tender offer and the closing of the proposed acquisition; the prospective benefits of the proposed acquisition, including expectations that it will strengthen BioMarin's enzyme therapies portfolio and be a strong strategic fit for BioMarin; Inozyme's product candidate INZ-701 and expectations regarding its ongoing development, including the potential for INZ-701 to be the first treatment for children and adults with ENPP1 Deficiency, the potential benefits of INZ-701 for patients, the anticipated timing for data from the first Phase 3 pivotal study of INZ-701 in children, the anticipated costs of developing INZ-701 and the potential regulatory approval of INZ-701 in 2027; potential revenue for INZ-701; additional INZ-701 clinical programs intended to expand to patients of all ages; the anticipated market for INZ-701; plans for an INZ-701 pivotal study for adolescents and adults; INZ-701's potential to expand into additional indications where deficiencies in the Pyrophosphate-Adenosine Pathway contribute to disease pathology; the accounting treatment of the potential acquisition under GAAP and its potential impact on BioMarin's financial results and financial guidance; BioMarin's plans for external innovation, including BioMarin being in a strong financial position to acquire additional assets; BioMarin's ability to execute additional transactions in future quarters; statements about BioMarin's future financial performance, including the expectations of Non-GAAP Operating Margin percentage; and other statements that are not historical facts. Actual results could differ materially from those anticipated in these forward-looking statements. Except as required by law, each of BioMarin and Inozyme assume no obligation to update these forward-looking statements, whether as a result of new information, future events or otherwise. These statements, which represent each of BioMarin's and Inozyme's current expectations or beliefs concerning various future events that are subject to significant risks and uncertainties, may contain words such as "may," "will," "would," "could," "expect," "anticipate," "intend," "plan," "believe," "estimate," "project," "seek," "should," "strategy," "future," "opportunity," "potential" or other similar words and expressions indicating future results.
These forward-looking statements are predictions and involve risks and uncertainties such that actual results may differ materially from these statements. Forward-looking statements reflect current beliefs and expectations; however, these statements involve inherent risks and uncertainties, including, without limitation, with respect to: consummating the proposed acquisition in the anticipated timeframe, if at all; how many of Inozyme's stockholders will tender their stock in the tender offer; the possibility that competing offers or acquisition proposals will be made; the possibility that various closing conditions for the transaction may not be satisfied or waived, including that a governmental entity may prohibit, delay, or refuse to grant approval for the consummation of the transaction (or only grant approval subject to adverse conditions or limitations); the difficulty of predicting the timing or outcome of regulatory approvals or actions, if any; the effects of the proposed acquisition (or the announcement thereof) on Inozyme's or BioMarin's stock price and/or BioMarin's or Inozyme's operating results; unknown or inestimable liabilities; the development, launch and commercialization of products and product candidates such as INZ-701, if approved; the successful completion of regulatory activities with respect to INZ-701; the parties' ability to realize the anticipated benefits of the proposed acquisition, including the possibility that the expected benefits from the proposed acquisition will not be realized or will not be realized within the expected time period and that BioMarin and Inozyme will not be integrated successfully or that such integration may be more difficult, time-consuming or costly than expected; obtaining and maintaining adequate coverage and reimbursement for BioMarin's or Inozyme's products; the time-consuming and uncertain regulatory approval process; the costly and time-consuming pharmaceutical product development process and the uncertainty of clinical success, including risks related to failure or delays in successfully initiating or completing clinical trials and assessing patients, including with respect to current and planned future clinical trials of INZ-701; global economic, financial, and healthcare system disruptions and the current and potential future negative impacts to BioMarin's or Inozyme's business operations and financial results; the sufficiency of BioMarin's or Inozyme's cash flows and capital resources; BioMarin's ability to fund the acquisition with existing cash and investments; BioMarin's evaluation of the accounting treatment of the potential acquisition and its potential impact on its financial results and financial guidance; BioMarin's or Inozyme's ability to achieve targeted or expected future financial performance and results and the uncertainty of future tax, accounting and other provisions and estimates; the effects of the transaction on relationships with key third parties, including employees, customers, suppliers, other business partners or governmental entities, including the risk that the proposed acquisition adversely affects employee retention; transaction costs; risks that the proposed acquisition disrupts current plans and operations; risks that the proposed transaction diverts management's attention from ongoing business operations; changes in Inozyme's business during the period between announcement and closing of the proposed acquisition; any legal proceedings and/or regulatory actions that may be instituted related to the proposed acquisition; and other risks and uncertainties affecting BioMarin and Inozyme, including those risk factors detailed in BioMarin's and Inozyme's filings with the Securities and Exchange Commission (SEC), including, without limitation, the risk factors contained under the caption "Risk Factors" in BioMarin's Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2025 and Inozyme's Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2025, as such risk factors may be updated by any subsequent reports, as well as the Tender Offer Statement on Schedule TO and related tender offer documents to be filed by BioMarin and its acquisition subsidiary, Incline Merger Sub, Inc., and the Solicitation/Recommendation Statement on Schedule 14D-9 to be filed by Inozyme. Stockholders of BioMarin and Inozyme are urged not to place undue reliance on forward-looking statements, which speak only as of the date hereof. BioMarin and Inozyme are under no obligation, and expressly disclaim any obligation, to update (publicly or otherwise) or alter any forward-looking statement, including without limitation any financial projection or guidance, whether as a result of new information, future events or otherwise.
BioMarin® is a registered trademark of BioMarin Pharmaceutical Inc. or its affiliates. Inozyme® is a registered trademark of Inozyme Pharma Inc. or its affiliates.
Additional Information about the Acquisition and Where to Find It
The tender offer for all of the outstanding shares of Inozyme described in this communication has not yet commenced. This communication is for informational purposes only and is neither an offer to purchase nor a solicitation of an offer to sell any securities, nor is it a substitute for the tender offer materials that Inozyme, BioMarin or its acquisition subsidiary will file with the SEC upon commencement of the tender offer. The solicitation and offer to tender and the offer to buy outstanding shares of Inozyme will only be made pursuant to a tender offer statement on Schedule TO, including an Offer to Purchase and related tender offer materials that BioMarin and its acquisition subsidiary, Incline Merger Sub, Inc., are expected to file with the SEC. At the time the tender offer is commenced, BioMarin and its acquisition subsidiary will file a Tender Offer Statement on Schedule TO, and Inozyme will file a Solicitation/Recommendation Statement on Schedule 14D-9 with the SEC with respect to the tender offer. THE TENDER OFFER MATERIALS (INCLUDING AN OFFER TO PURCHASE, A RELATED LETTER OF TRANSMITTAL AND CERTAIN OTHER TENDER OFFER DOCUMENTS), AS WELL AS THE SOLICITATION/RECOMMENDATION STATEMENT ON SCHEDULE 14D-9, WILL CONTAIN IMPORTANT INFORMATION ABOUT THE PROPOSED ACQUISITION AND THE PARTIES THERETO. INVESTORS AND STOCKHOLDERS OF INOZYME ARE URGED TO READ THESE DOCUMENTS CAREFULLY WHEN THEY BECOME AVAILABLE (AND EACH AS IT MAY BE AMENDED OR SUPPLEMENTED FROM TIME TO TIME) BECAUSE THEY WILL CONTAIN IMPORTANT INFORMATION THAT INVESTORS AND STOCKHOLDERS OF INOZYME SHOULD CONSIDER BEFORE MAKING ANY DECISION WITH RESPECT TO THE TENDER OFFER. The tender offer materials (including the Offer to Purchase, the related Letter of Transmittal and certain other tender offer documents), as well as the Solicitation/Recommendation Statement, will be made available to all investors and stockholders of Inozyme at no expense to them at SEC's website at www.sec.gov. Copies of the documents filed with the SEC by BioMarin will be available free of charge on BioMarin's website at www.biomarin.com. Copies of the documents filed with the SEC by Inozyme will be available free of charge on Inozyme's website, www.inozyme.com, or by contacting Inozyme's investor relations department at investorrelations@inozyme.com. The information contained in, or that can be accessed through, BioMarin's and Inozyme's websites is not a part of, or incorporated by reference herein. In addition to the Offer to Purchase, related Letter of Transmittal and certain other tender offer documents, and Solicitation/Recommendation Statement, BioMarin and Inozyme file annual, quarterly, and current reports, proxy statements and other information with the SEC. You may read any reports, statements or other information filed by BioMarin and Inozyme with the SEC for free on the SEC's website at www.sec.gov.
BioMarin Contacts
Traci McCarty, Investors BioMarin Pharmaceutical Inc.
(415) 455-7558
Marni Kottle, Media
BioMarin Pharmaceutical Inc.
(415) 218-7111
Inozyme Contacts
Stefan Riley, Investors Inozyme Pharma, Inc. (617) 461-2442
Todd Cooper, Media Biongage Communications (617) 840-1637
SOURCE BioMarin Pharmaceutical Inc.
- Angelini Ventures and Apollo Health Ventures join existing top-tier investor syndicate
- Follows original $36M Series A raise and brings Series A total to $75M
- Proceeds are being used to fund Phase 1b Alzheimer’s and DME studies, and bispecific development
Sacramento, May 14, 2025 (GLOBE NEWSWIRE) - Therini Bio, Inc., a clinical-stage biotech company developing fibrin-targeting immunotherapies for neurodegenerative diseases driven by vascular dysfunction today announced that it raised $39 million in a Series A extension financing. The extension financing included new investors, Angelini Ventures and Apollo Health Ventures, in addition to existing investors SV Health Investors’ Biotech Fund and Dementia Discovery Fund, MRL Ventures Fund, Sanofi Ventures, Eli Lilly and Company, Dolby Family Ventures and Foundation for a Better World. This financing increased Therini Bio’s Series A total to $75M.
Therini Bio anticipates using the proceeds to fund Phase 1b trials evaluating its lead candidate, THN391, for the treatment of Alzheimer’s Disease (AD) and Diabetic Macular Edema (DME), as well as support development of a fibrin/VEGF bispecific.
Therini Bio seeks to target the underlying causes of neurodegeneration. Factors such as aging, genetics and prevalent diseases, such as hypertension and diabetes, lead to vascular dysfunction and the accumulation of toxic fibrin deposits outside of blood vessels. These deposits induce chronic neuroinflammation, resulting in neuronal damage and severe neurogenerative diseases. Therini Bio has developed a novel approach aimed at treating these devastating diseases by specifically targeting the inflammatory epitope on fibrin to halt the destructive effects of neuroinflammation.
THN391, a potential first-in-class, high-affinity humanized monoclonal antibody designed to selectively block fibrin-mediated neuroinflammation without affecting coagulation pathways, demonstrated activity in preventing vascular and neuronal degeneration in preclinical studies of AD and retinal diseases. In a recent Phase 1a trial, THN391 was well-tolerated in healthy volunteers, showed no adverse hematological effects or impact on coagulation and fibrinolysis, lacked an anti-drug antibody response and had dose-proportional pharmacokinetics with a half-life supporting monthly dosing. Supported by the current data and financing, the experienced team at Therini Bio is preparing to begin dosing patients in Phase 1b trials evaluating THN391 in AD and DME.
Tara Nickerson, Ph.D., Chief Executive Officer of Therini Bio and an industry veteran who also played an instrumental role previously in several prominent private and public biotech financings, including as Chief Business Officer of Maze Therapeutics and Prothena Biosciences, remarked, “We are deeply grateful to partner with such a distinguished investor group, both new and old. Their support enables us to significantly advance our shared vision of delivering patients a rational, innovative approach to potentially treat their debilitating conditions, including Alzheimer’s disease and Diabetic Macular Edema. We look forward to advancing the Phase 1b trials to demonstrate the potential benefit of this novel mechanism in patients.”
Thomas Thestrup, Senior Principal at Angelini Ventures, who led the investment on behalf of Angelini Ventures stated, ”We are proud to support Therini Bio’s mission to address the underlying vascular and inflammatory drivers of neurodegenerative disease through a truly novel approach. As an active CNS investor, we are excited about Therini’s first-in-class selective antibody therapy targeting fibrin-mediated inflammation, offering a groundbreaking path to transform the treatment of diseases such as Alzheimer’s and DME.”
About
Therini Bio is a clinical-stage biotech company developing immunotherapies for neuroinflammation in diseases driven by vascular dysfunction. Therini Bio is developing a pipeline of potential first-in-class therapies selectively targeting toxic fibrin accumulation for diseases, including Alzheimer’s disease and Diabetic Macular Edema, where destructive neuroinflammation plays a central role in the disease process. Therini Bio’s top-tier syndicate of life sciences investors includes the Alzheimer’s Drug Discovery Foundation, Angelini Ventures, Apollo Health Ventures, SV Health Investors’ Biotech Fund and Dementia Discovery Fund, Dolby Family Ventures, Dreavent Biotech Investments, Eli Lilly and Company, Foundation for a Better World, MRL Ventures Fund, the therapeutics-focused corporate venture fund of Merck & Co., Inc., and Sanofi Ventures. For more information, visit www.therinibio.com.
Contact
Tara Nickerson, Ph.D.
Chief Executive Officer
tnickerson@therinibio.com
- Novel approach to treat neurodegenerative diseases by targeting fibrin-driven neuroinflammation was well-tolerated among healthy volunteers, with a clean hematological profile -
- Half-life supports once-monthly dosing -
- Data to be presented at AAIC 25-
Sacramento, April 29, 2025 (GLOBE NEWSWIRE) – Therini Bio, Inc., a clinical-stage biotech company developing fibrin-targeting immunotherapies for neurodegenerative diseases driven by vascular dysfunction today announced positive results from a Phase 1a trial assessing its lead candidate, THN391, in healthy volunteers for the treatment of neurodegenerative diseases. The data from this trial, which showcases Therini’s novel approach to treating neurodegenerative diseases, will be presented at the Alzheimer's Association International Conference 2025 (AAIC 25) in Toronto, Canada, on Wednesday, July 30, 2025.
THN391 is a potential first-in-class, high-affinity, humanized monoclonal antibody that is designed to selectively block fibrin-mediated neuroinflammation without interfering with coagulation pathways. At sites of vascular dysfunction, deposited fibrin binds complement receptors on innate immune cells triggering inflammation and neuronal damage in the brain and retina. In preclinical studies of Alzheimer’s disease and retinal diseases, antibodies blocking fibrin inflammation demonstrated effectiveness in protecting against vascular and neuronal degeneration.
The Phase 1a, randomized, double-blind, placebo-controlled trial assessed the safety, tolerability and pharmacokinetics (PK) of single and multiple ascending doses of THN391 in healthy volunteers. THN391 was well-tolerated, with no Serious Adverse Events reported. THN391 also demonstrated a clean hematological profile, with no impact on coagulation and fibrinolysis, and did not induce an anti-drug antibody response. Additionally, THN391 exhibited dose proportional PK and a half-life that supports monthly dosing.
“The results of this trial mark an important milestone for a new class of drugs for the treatment of neurodegenerative diseases,” said Tara Nickerson, Ph.D., Chief Executive Officer of Therini Bio. “By targeting vascular dysfunction and chronic neuroinflammation, we aim to address fundamental root causes of neurodegeneration. Galvanized by the encouraging data and compelling preclinical evidence, we are eager to accelerate the development of THN391 to potentially ameliorate the lives of patients devastated by debilitating diseases, including Alzheimer’s and Diabetic Macular Edema.”
Therini Bio intends to commence two Phase 1b trials imminently, evaluating THN391 for the treatment of patients with Alzheimer’s Disease and Diabetic Macular Edema.
About
Therini Bio is a clinical-stage biotech company developing immunotherapies for neuroinflammation in diseases driven by vascular dysfunction. Therini is developing a pipeline of potential first-in-class therapies selectively targeting toxic fibrin accumulation for diseases, including Alzheimer’s disease (AD) and Diabetic Macular Edema (DME), where destructive neuroinflammation plays a central role in the disease process. Therini Bio’s top-tier syndicate of life sciences investors includes the Alzheimer’s Drug Discovery Foundation, SV Health Investors’ Biotech Fund and Dementia Discovery Fund, Angelini Ventures, Apollo Health Ventures, Dolby Family Ventures, Dreavent Biotech Investments, Eli Lilly and Company, Foundation for a Better World, MRL Ventures Fund, the therapeutics-focused corporate venture fund of Merck & Co., Inc., and Sanofi Ventures. For more information, visit www.therinibio.com.
Contact
Tara Nickerson, Ph.D.
Chief Executive Officer
tnickerson@therinibio.com

-Clinical-stage company targeting fundamental drivers of inflammation, autoimmunity and fibrosis, founded by Versant Ventures and Novartis Venture Fund-
-Series B financing led by Forbion and Sanofi Ventures-
-Antibody programs developed in collaboration with Versant’s Ridgeline Discovery Engine in Basel-
Basel, Switzerland and San Francisco, Calif., April 24, 2025 - Granite Bio AG, a clinical-stage immunology company, has emerged from stealth with $100 million in funding. This includes a $30 million Series A led by founding investors Versant Ventures and Novartis Venture Fund, and a $70 million Series B led by Forbion and Sanofi Ventures.
Granite’s pipeline features two first-in-class antibodies targeting multiple autoimmune diseases that address large market opportunities.
GRT-001 depletes pro-inflammatory monocytes, key drivers of autoimmunity and inflammation. In non-human primate studies, GRT-001 efficiently and dose-dependently depleted pro-inflammatory monocytes. Treatment was well tolerated and spared tissue-resident macrophages, which are important for tissue homeostasis. GRT-001 is currently in Phase 1a testing in healthy volunteers and is expected to enter a Phase 1b trial in patients with inflammatory bowel disease later this year.
GRT-002 blocks interleukin-3, a key player in autoimmune inflammation and type II inflammation, offering a new approach to treating itch and allergy. The molecule is in preclinical development and is expected to enter clinical trials in 2026.
Both of Granite’s lead molecules were developed in collaboration with Versant’s Ridgeline Discovery Engine in Basel, Switzerland, and originated from the laboratories of Professor and Scientific Co-Founder Matthias Mack at University of Regensburg.
"Granite is pioneering a new approach to tackling inflammation, autoimmunity, and fibrosis by addressing fundamental disease drivers at their source,” said Patrick Loustau, president and CEO. “With the support of an exceptional investor syndicate and a world-class team, we are advancing a pipeline of first-in-class therapies with the potential to transform patient outcomes. I look foward to working with the management team to deliver groundbreaking treatments that address the urgent unmet needs in immunology and beyond."
“Despite multiple immunology-based therapeutic approaches currently commercialized, patients with inflammatory disorders continue to experience a lack of symptom control and relapse,” said Nigel Sheail, partner at Versant and Granite board member. “Granite has the potential to enable a real step change for patients with its innovative programs that target key fundamental disease pathways.”
“I am proud to support Granite as it advances its lead assets using a truly differentiated approach to tackling inflammation at its root,” said Rogier Rooswinkel, Ph.D., General Partner at Forbion and Granite board member. “With the strength of this investor syndicate and an exceptional leadership team, Granite is well-positioned to drive its pipeline forward and deliver meaningful new therapies for patients.”
Leadership team
Granite is led by an experienced team including:
- Patrick Loustau, president and CEO, brings broad and successful experience in building and developing organizations in biotech and pharmaceutical companies. Prior to joining Granite Bio, he was the CBO of Amolyt Pharma until its acquisition by Alexion (AstraZeneca Rare Disease) for up to $1.05B in July 2024. Before Amolyt, he was CEO of Zumbro Discovery Inc. and president of Zafgen Inc. Earlier in his career, he led global organizations at Bristol Myers-Squibb and Novo Nordisk.
- Dominik Hartl, M.D., CMO has extensive background in immunology and fibrosis drug development. He previously served as CMO for Quell Therapeutics, a company specialized in immune-mediated diseases. Before that, he held leadership positions in Immunology Drug Development/Translational Medicine at Novartis (NIBR/Immunology), Roche (pRED/I2O) and Galapagos Pharma. Dominik is a board-certified M.D. and holds an adjunct professorship in Pediatric Immunology at University of Tübingen.
- Gijs van den Brink, M.D., Ph.D., CSO, possesses deep academic and industry experience in immunology and IBD. Currently also an Operating Partner at Forbion, he previously served as SVP and Global Head of Immunology, CVM, ID, and Ophthalmology Discovery and Early Development at Roche. Before that, Dr. van den Brink was head of Immunology discovery and early and late-stage clinical development at GlaxoSmithKline plc. Gijs previously was a gastroenterologist and professor of experimental gastroenterology and co-authored over 150 peer-reviewed scientific publications.
- Eliot Forster, chairperson of the board, has more than 30 years of extensive biotech experience from both executive and non-executive roles. He was CEO of F-star Therapeutics until its acquisition by inovX Pharma LTD in March 2023, and formerly CEO of Immunocore. Eliot is currently CEO of Levicept LTD, non-executive chair of Tessellate BIO and a director at Immatics.
About Granite Bio
Granite Bio is a biotechnology company developing first-in-class antibodies that target the root causes of a variety of inflammatory, autoimmune and fibrotic conditions. Granite is backed by leading healthcare venture firms including Versant Ventures, Novartis Venture Fund, Forbion and Sanofi Ventures. For more information, please visit www.GRANITEBIO.com.
About Versant Ventures
Versant Ventures is a leading healthcare venture capital firm committed to helping exceptional entrepreneurs build the next generation of great companies. The firm’s emphasis is on biotechnology companies that are discovering and developing novel therapeutics. With $5.3 billion under management and offices in the U.S., Canada and Europe, Versant has built a team with deep investment, operating and R&D expertise that enables a hands-on approach to company building. Since the firm’s founding in 1999, more than 95 Versant companies have achieved successful acquisitions or IPOs. For more information, please visit www.versantventures.com.
About NVF
Novartis Venture Fund is a financially driven corporate life science venture fund whose purpose is to foster innovation, drive significant patient benefit and generate superior returns by creating and investing in innovative life science companies at various stages of their development. For more information, go to www.nvfund.com.
About Forbion
Forbion is a leading global venture capital firm with deep expertise in Europe and offices in Naarden, The Netherlands, Munich, Germany and Boston, USA. Forbion invests in innovative biotech companies, managing approximately €5 billion across multiple fund strategies that cover all stages of (bio-) pharmaceutical drug development. In addition, Forbion leverages its biotech expertise beyond human health to address ‘planetary health’ challenges through its BioEconomy fund strategy, which invests in companies developing sustainable solutions in food, agriculture, materials, and environmental technologies. Forbion’s team consists of over 30 investment professionals that have built an impressive performance track record since the late nineties with 128 investments across 11 funds. Forbion’s record of sourcing, building and guiding life sciences companies has resulted in many approved breakthrough therapies and valuable exits. Forbion typically selects impactful investments that will positively affect the health and well-being of people and the planet, as well as meet its financial return objectives. The firm is a signatory to the United Nations Principles for Responsible Investment. Forbion operates a joint venture with BGV, the manager of seed and early-stage funds, especially focused on Benelux and Germany.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi, focused on investing in promising early-stage healthcare companies. The firm supports pioneering innovations in biotechnology, digital health, and life sciences aligning with Sanofi’s mission to bring life-changing treatments to patients worldwide. For more information, please visit www.sanofiventures.com.
Granite Bio contact:
Steve Edelson
415-801-8088

- GLM101 is the first disease-modifying therapeutic in development to treat PMM2-CDG, the most common congenital disorder of glycosylation
- Data from the ongoing Phase 2 open-label study of GLM101 provide clinical proof of concept, showing improvement in ataxia, a key burden of disease in PMM2-CDG
- Funding will support advancement of GLM101 into a randomized, placebo-controlled Phase 2b safety and efficacy study
SAN CARLOS, Calif., April 16, 2025 – Glycomine, Inc., a biotechnology company focused on developing transformative new therapies for orphan diseases, today announced a $115 million Series C financing to advance its lead candidate, GLM101, into a Phase 2b clinical trial. The financing was led by CTI Life Sciences Fund, funds managed by abrdn Inc., and Advent Life Sciences, alongside continued investment from existing investors, Novo Holdings, Sanofi Ventures, Abingworth, RiverVest Venture Partners, Sanderling Ventures, Chiesi Ventures, Remiges Ventures, and Asahi Kasei Ventures.
“We are excited to partner with our new investors who have strong track records in rare diseases and for the continued support from our existing investors,” said Steve Axon, Glycomine’s CEO. “This financing will enable us to advance GLM101 into a randomized, placebo-controlled trial later this year—an important step toward bringing the first disease-modifying therapeutic to patients with PMM2-CDG.”
GLM101, a first-in-class mannose-1-phosphate replacement therapy, is in development for phosphomannomutase-2 congenital disorder of glycosylation (PMM2-CDG), a rare and life-threatening genetic disorder with no approved treatments. Glycomine has enrolled more than 20 patients across Europe and the U.S. in its ongoing Phase 2 study and recently initiated dosing in pediatric patients.
Data from Glycomine’s ongoing Phase 2 open-label study have demonstrated promising improvements in ataxia, a hallmark debilitating manifestation of PMM2-CDG. Among nine adult and adolescent patients, treatment with GLM101 led to an average 11.9-point improvement on the ICARS (International Cooperative Ataxia Rating Scale) over 24 weeks.
“We are impressed by the therapeutic approach and strong progress of the Glycomine team,” said Youssef Bennani, Ph.D., Managing Partner with CTI Ventures. “We are excited to be part of such a strong investor syndicate and look forward to the potential of making a positive impact in the lives of patients with PMM2-CDG.”
Dominic Schmidt, Ph.D., General Partner with Advent Life Sciences added, “We are highly encouraged by the clinical signal observed in the ongoing Phase 2 study. Most notably, the data show strong potential for clinically meaningful improvement in ataxia, a key driver of disease burden for PMM2-CDG patients.”
In connection with the financing, Drs. Bennani and Schmidt have been appointed to Glycomine’s Board of Directors.
About PMM2-CDG
Phosphomannomutase 2-congenital disorder of glycosylation (PMM2-CDG), previously known as CDG-1a, is the most prevalent congenital disease of glycosylation. PMM2-CDG is caused by a genetic mutation in phosphomannomutase 2 (PMM2), which results in the protein having reduced activity. PMM2 is an enzyme that converts mannose-6-phosphate to mannose-1-phosphate, which is required to insert the mannose sugar building block into developing glycans that are crucial for proper protein structure and function. The deficiency of mannose-1-phosphate disrupts the process of N-glycosylation and causes a wide array of clinical symptoms and, in many cases, can be life-threatening.
About Glycomine, Inc.
Glycomine is a clinical-stage biotechnology company that is advancing treatments for serious rare diseases for which no other therapeutic options exist. The Company’s lead investigational drug candidate GLM101 is a mannose-1-phosphate replacement therapy in development to treat PMM2-CDG. GLM101 is designed to deliver mannose-1-phosphate into cells and thereby bypass disease-causing PMM2 mutations to restore pathway function. GLM101 has received Orphan Drug Designation in the U.S. and E.U. and Rare Pediatric Disease Designation and Fast Track Designation in the U.S. The company is based in San Carlos, California, and supported by leading international life sciences investors. For more info visit www.glycomine.com.
Corporate Contact:
Peter McWilliams, Ph.D.
info@glycomine.com
Media Contact:
Jessica Yingling, Ph.D.
Little Dog Communications Inc.
jessica@litldog.com

- Developed by Click Therapeutics, CT-132 underwent FDA review via the De Novo pathway for first-in-class medical devices as the first digital therapeutic for the preventive treatment of episodic migraine in the United States.
- FDA marketing authorization for CT-132 is supported by data from both the pivotal ReMMi-D randomized controlled trial, in which CT-132 met its primary endpoint by significantly reducing monthly migraine days on top of background pharmacotherapy, and the bridging study, ReMMiD-C, where CT-132 showed similar performance in patients on calcitonin gene-related peptide (CGRP) inhibitors.
- CT-132 studies may provide data to inform future development of added clinical benefit claims consistent with the Prescription Drug Use-Related Software (PDURS) FDA Draft Guidance.
NEW YORK, N.Y. (April 15, 2025) – Click Therapeutics, Inc., (“Click”) a leader in prescription medical treatments as both prescription digital therapeutics and software-enhanced drug™ therapies, has obtained FDA marketing authorization for the first prescription digital therapeutic for the preventive treatment of episodic migraine. FDA granted the De Novo Classification Request for the company’s prescription digital therapeutic, CT-132, for the preventive treatment of episodic migraine in patients 18 years of age and older. It is intended for adjunctive use alongside acute and/or other preventive treatments for migraine. The marketing authorization for CT-132 is based on data from the ReMMi-D (Reduction in Monthly Migraine Days, NCT05853900) study in patients taking standard-of-care prescription migraine medications (acute, preventive first-line, and preventive second-line), where CT-132 met its primary endpoint. Clinical results from the ReMMiD-C bridging study (NCT06004388), which showed that CT-132 performed similarly in patients taking calcitonin gene-related peptide (CGRP) inhibitors, were also submitted in the De Novo and included in the product’s FDA premarket review.
“This marks a significant milestone for the more than 37 million adults in the US who live with migraine,” said Shaheen Lakhan, MD, PhD, FAAN, chief medical and scientific officer of Click Therapeutics. “As a groundbreaking digital therapeutic for migraine prevention, CT-132 offers eligible patients a new path to reducing the burden caused by migraine, one they can access anywhere via an evidence-based mobile application on their smartphone, significantly improving accessibility and expanding care to patients.”
Click Therapeutics recently presented CT-132 pivotal study results at the American Academy of Neurology Annual Meeting, where co-investigator Stewart J. Tepper, MD, Vice President of the New England Institute for Neurology and Headache and scientific advisor to Click shared in a post-conference Medscape article, “I think this is very exciting as a clinician who takes care of patients because we don’t have anything like this in our migraine armamentarium. We know that behavioral techniques are helpful adjunctively, but large areas of the country just don’t have access to them.”
“With this landmark, first-in-class FDA authorization in episodic migraine, Click’s interventions have now demonstrated clinically meaningful benefit across three unique therapeutic areas, including psychiatry, cardiometabolic disease and now neurology,” said David Benshoof Klein, chief executive officer of Click Therapeutics. “As the first authorization in our neurology pipeline, and the first of our PDTs to target and successfully treat a pain-related condition, it confirms the power of Click’s platform to deliver meaningful outcomes across therapeutic areas.”
Built on Click’s industry-leading AI-enabled platform, CT-132 combines scientifically proven therapies with proprietary mechanisms of action to deliver clinically-meaningful interventions for patients with episodic migraine. Click Therapeutics designs patient-centric applications like CT-132 by incorporating storytelling, user research, and elements of consumer technology to increase engagement and drive improved clinical outcomes, with the goal of delivering personalized treatment for effective migraine management.
Intended for adjunctive use alongside other treatments for migraine, CT-132 demonstrated in clinical testing the ability to add clinically meaningful benefit on top of background pharmacotherapy. CT-132 is thus well-positioned for further development in the future as a software-enhanced drug™ therapy. Click’s software-enhanced drug therapies combine software with pharmacotherapy to create software-enhanced drug™ treatment options, targeting the unique needs of a specific medication to deliver added clinical benefit to patients. Click launched its new product offering, Click SE™, in October 2024 to pioneer this new therapeutic category in response to increasing interest in the U.S. Food and Drug Administration (FDA) draft guidance on Prescription Drug Use-Related Software (PDURS).
This marketing authorization announcement follows Click's recent Series C funding round news, announced in March 2025.
Indication
The CT-132 prescription digital therapeutic is indicated for the preventive treatment of episodic migraine in patients 18 years of age and older. It is intended for adjunctive use alongside acute and / or other preventive treatments for migraine.
Safety Information:
There are no contraindications to using CT-132. CT-132 is not intended to be used as a standalone therapy. CT-132 does not replace or substitute other migraine treatments, including medication for migraine. Patients should continue their current treatment as directed.
About the ReMMi-D Pivotal Study and ReMMiD-C Bridging study
The indications for use for CT-132 are supported by the results from two double-blind decentralized randomized controlled trials, a pivotal study (ReMMi-D) and a bridging study (ReMMiD-C). The pivotal ReMMi-D study evaluated the effectiveness and safety of CT-132 for the prevention of episodic migraine in patients 18 years and older, compared to a sham digital control and included patients (n=558) on the most commonly prescribed migraine medications. ReMMiD-C was designed identically to ReMMi-D but required that participants (n=110) be taking at least one calcitonin gene-related peptide (CGRP) inhibitor, though participants were also permitted use of non–migraine-specific acute or preventive medications. The study designs mirrored those of contemporary randomized control trials for migraine drugs. However, unlike in drug studies, patients continued their existing migraine medications without any washout period, so the additive treatment effect of CT-132 on top of background pharmacotherapy could be evaluated.
Both studies demonstrated that CT-132 reduced monthly migraine days (MMDs) compared to a sham digital control in patients already using acute and preventive migraine medications, with no device-related adverse events reported. At the completion of the pivotal ReMMi-D study, CT-132 demonstrated a statistically significant reduction in monthly migraine days (MMDs) after 12 weeks of treatment compared to the sham digital control (n=568, ITT population; treatment difference: –0.9 MMDs; p=0.005). Participants in the treatment arm experienced a mean reduction of –3.04 MMDs by the end of the intervention. Further, migraine related quality of life, assessed by the Migraine-Specific Quality-of-Life Questionnaire (MSQ, revealed a difference between the CT-132 and sham digital control arms from as early as 4 weeks and through weeks 8 and 12. Migraine disability, as measured by the Migraine Disability Assessment (MIDAS) score, improved more in the CT-132 arm than in the sham arm at the end of treatment. Note that the analysis of secondary effectiveness endpoints did not include multiplicity adjustment or formal hypothesis testing. Engagement and adherence were high and sustained over the full 12-week duration of the treatment, with a median of 81 daily lessons out of 84 completed by those receiving the sham digital control and a median of 84 completed by those receiving CT-132. CT-132 was well-tolerated, with no discontinuations due to treatment-emergent adverse events (TEAEs). The ReMMiD-C bridging study provided supportive information, also evaluated versus a sham digital control but without formal hypothesis testing, that demonstrated similar performance of CT-132 in patients taking the new-class of migraine-specific medications, CGRP inhibitors.
About Migraine
Migraine is a complex and debilitating condition that affects more than 37 million adults and is the second leading cause of disability in the United States.1,2 It is characterized by episodes of moderate-to-severe headache and is generally associated with nausea and increased sensitivity to light and sound.3 For those living with migraine, attacks unfold over hours to days and negatively impact key domains of life including employment, educational attainment, and relationships.4 Despite the availability of preventive migraine medications, there remains a significant unmet need in migraine management. Many patients continue to experience frequent and debilitating attacks, even when using these drugs as prescribed.5
About Prescription Digital Therapeutics
Prescription Digital Therapeutics (PDTs) deliver evidence-based treatments directly to a patient via their smartphone in the form of a mobile app. With PDTs, the software is the treatment. PDTs are prescribed by a physician to treat a disease or condition, and as such are clinically validated and FDA-regulated. PDTs can be used independently or in combination with traditional pharmaceutical treatments.
About Click Therapeutics
Click Therapeutics, Inc. develops, validates, and commercializes software as prescription medical treatments for people with unmet medical needs. We are expanding the possibilities of medicine with Digital Therapeutics™ that combine clinical science with the power of software to create a new way to treat disease.
Operating at the intersection of biology and technology, we use a proprietary platform-based approach to therapeutic development that leverages patient-centric design principles and innovative AI-based technologies to deliver a unique combination of engagement and clinical outcomes, consistently. Digital therapeutics on Click’s platform are regulated, clinically validated prescription mobile applications that are being developed to address diverse areas of therapeutic need, including indications in psychiatry, neurology, oncology, immunology, and cardiometabolic diseases. In 2024, in response to FDA guidance on prescription drug use-related software (PDURS) and building off the capabilities of our platform, we launched Click SE™ to extend our digital therapeutics platform and expertise to the development of software-enhanced drug™ therapies that combine software with pharmacotherapy to offer added clinically meaningful benefit to patients.
To date, three Click Therapeutics devices have received FDA marketing authorizations. The company's self-developed CT-132, a first-in-class prescription digital therapeutic for preventing episodic migraine, was granted De Novo classification by the FDA. Click Therapeutics, in collaboration with Otsuka, developed Rejoyn™, which became the first prescription digital therapeutic authorized by the FDA for the adjunctive treatment of major depressive disorder symptoms. Additionally, the company expanded its portfolio into cardiometabolic disease with AspyreRx, which received FDA marketing authorization for the treatment of type 2 diabetes.
Our commitment to advancing digital medicine means we continually improve our platform technologies, ensuring we stay at the forefront of cognitive, behavioral, and neuromodulatory therapeutic innovation, to achieve the best possible outcomes for patients. Our diverse team of innovators—spanning clinicians, researchers, technologists, designers and more—works together to create cutting-edge digital therapeutics, united in the mission to transform patient care. For more information, visit www.clicktherapeutics.com and connect with us on LinkedIn.
References
- American Migraine Foundation https://americanmigrainefoundation.org/resource-library/migraine-facts/
- Steiner TJ, et al. J Headache Pain. 2020;21(1):137. https://pubmed.ncbi.nlm.nih.gov/33267788/
- National Institute of Neurological Disorders and Stroke https://www.ninds.nih.gov/health-information/disorders/migraine
- Buse, D. C., Fanning, K. M., Reed, M. L., Murray, S., Dumas, P. K., Adams, A. M., & Lipton, R. B. (2019). Life With Migraine: Effects on Relationships, Career, and Finances From the Chronic Migraine Epidemiology and Outcomes (CaMEO) Study. Headache, 59(8), 1286–1299. https://doi.org/10.1111/head.13613
- Bentivegna, E., Onan, D., & Martelletti, P. (2023). Unmet Needs in Preventive Treatment of Migraine. Neurology and therapy, 12(2), 337–342. https://doi.org/10.1007/s40120-023-00438-z
Series C round led by Deep Track Capital with participation fromadditional new and existing investors
Financing will fund early-to-mid-stage clinical studies for ATTO-1310 inchronic pruritus and ATTO-3712 in atopic dermatitis, and multiple earlier-stage multi-specific programs
SAN CARLOS, Calif., April 15, 2025 -- AttoviaTherapeutics, a privately-held, clinical-stage biopharma companydeveloping treatments for immune-mediated diseases with high unmetpatient need, today announced the close of a $90 million Series Cfinancing. The funding round was led by Deep Track Capital withparticipation from new investors including Vida Ventures, Sanofi Ventures,and Mirae Asset Capital Life Science, and ongoing support from existinginvestors including Frazier Life Sciences, venBio, Goldman SachsAlternatives, Nextech Ventures, Cormorant Asset Management, EcoR1Capital, Marshall Wace, and Illumina Ventures. In conjunction with thefinancing, Rebecca Luse, Managing Director at Deep Track Capital, will jointhe Company's Board of Directors.
Proceeds from the financing, along with the Company’s existing cash andinvestments, will be used to advance Attovia’s lead assets, ATTO-1310 andATTO-3712, through clinical proof-of-concept, with the aim of achievingbest-in-disease efficacy for the treatment of chronic pruritus and atopicdermatitis. The funds will also enable focused expansion of Attovia’s pipeline of multi-specific therapeutic candidates utilizing the company’sproprietary ATTOBODY technology to develop breakthrough treatmentoptions for patients suffering from immune-mediated disorders, includinginflammatory bowel disease (IBD) and others.
“Completing what was an oversubscribed round of financing during thistime speaks to the standout potential of our platform, programs, andexcellent team,” said Tao Fu, Attovia’s founder and CEO. “We deeplyappreciate the support of our exceptional investor base and the sharedcommitment to transforming the lives of patients living with immune-mediated diseases. We now have a multi-year runway to advance ourprograms expeditiously, expand our pipeline judiciously, and pursuepotential partnerships, all of which should give us greater optionality forthe future.”
The financing allows Attovia to continue to advance its first ATTOBODY-based product candidate, ATTO-1310, a first-in-class, half-life extended anti-IL31 biologic, currently in Phase 1 studies, to provide a highly efficacious,fast acting, and convenient treatment option for chronic pruritus ofunknown origin (CPUO)—a disease affecting millions of patients in the U.S.and other developed countries with currently no approved treatments—aswell as additional pruritic conditions with urgent unmet need.
Attovia is also advancing ATTO-3712, a first-in-class anti-IL13 x IL31, half-lifeextended bispecific designed to provide breakthrough efficacy andconvenient dosing in atopic dermatitis and potentially chronicspontaneous urticaria, prurigo nodularis, and other inflammatory skinconditions. The company plans to start Phase 1 clinical studies with ATTO-3712 in the second half of 2025.
Attovia’s earlier-stage pipeline includes ATTO-004, a multi-specific drugcandidate targeting IBD, and two other multi-specific discovery-stageprograms.
“Attovia has created a transformative pipeline leveraging its revolutionaryATTOBODY platform,” said Rebecca Luse, Managing Director at Deep TrackCapital. “We are highly impressed with the team's speed and quality ofdisciplined execution, and we are looking forward to supporting theirgrowth and success as they work to realize the full potential of theirplatform and pipeline programs.”
About Attovia
Attovia is creating a pipeline of biotherapeutics for the treatment ofimmune-mediated diseases. We leverage ATTOBODY™, our proprietarybiologics platform, to generate potentially first-in-class and best-in-classmulti-specifics.
ATTOBODIES utilize spatial positioning technology to achieve biparatopictarget engagement. The biparatopic binding mode of ATTOBODIES resultsin ultra-high affinity, which translates to best-in-class potency in biologicactivity. ATTOBODIES offer unconstrained engineering, making them anideal modality for the development of multi-specific biologics, with tunablehalf-life translating to the potential for quarterly or less frequent dosing inpatients. The high-throughput, evolution-based ATTOBODY discoveryplatform delivers a high degree of diversity at industry-leading speed,accelerating and de-risking therapeutics development, offering thepotential to become the next-generation modality of choice. Learn more atwww.attovia.com and follow us on LinkedIn.
Press Contact
Attovia Therapeutics:
info@attovia.com
Results demonstrated statistically significant effects on biomarkers that predict ALS disease progression and severity
Evidence of brain penetration, target engagement, and potential anti-seizure effects demonstrated through biomarkers for TMS-EMG and EEG
Safety and tolerability profile consistent with previously reported results for QRL-101
QurAlis intends to advance QRL-101 into proof-of-concept studies as a potentially best-in-class treatment for both ALS and epilepsy
CAMBRIDGE, Mass., March 12, 2025 – QurAlis Corporation (“QurAlis”), a clinical-stage biotechnology company driving scientific breakthroughs into powerful precision medicines that have the potential to alter the trajectory of neurodegenerative and neurological diseases, today announced positive topline data from QurAlis' Phase 1 proof-of-mechanism (PoM) clinical trial of QRL-101 in healthy volunteers evaluating biomarkers related to amyotrophic lateral sclerosis (ALS) and epilepsy.
Topline results demonstrated a dose-dependent, statistically significant decrease in motor nerve excitability threshold tracking (mNETT) strength-duration time constant (SDTC), the co-primary endpoint of the trial related to ALS. SDTC is a well-established electrophysiological biomarker of peripheral motor excitability related to ALS, where elevation of SDTC has been clinically shown to predict faster disease progression and mortality. The decrease in SDTC observed with QRL-101 in this study was approximately 50% greater than previously reported for the Kv7 potassium channel opener ezogabine in a single-dose study in people with ALS. Additional observations for multiple secondary and exploratory endpoints related to motor nerve excitability were also significantly impacted.
The clinical trial demonstrated a statistically significant impact on multiple secondary and exploratory endpoints related to epilepsy. These included transcranial magnetic stimulation electromyography (TMSEMG) intracortical facilitation which indicates effective inhibition of cortical excitability, as well as significant increases in electroencephalography (EEG) spectral power in beta and gamma spectral bands further demonstrating cortical target engagement and brain penetration. There was no statistically significant impact on the slower delta and theta EEG bands, which suggests a low potential for GABA-A receptor activation and sedation. The study did not reach statistical significance on the co-primary endpoint of TMS-EMG motor evoked potential (MEP) amplitude, another measure of corticospinal excitability.
Results also demonstrated that the safety and tolerability profile of QRL-101 was consistent with previously reported study results evaluating QRL-101 to date. There were no serious adverse events or discontinuations due to adverse events reported observed in the study.
“We are excited by these topline data from our biomarker study in healthy participants which suggest that QRL-101 has the potential to provide a therapeutic effect for both ALS and epilepsy,” said Kasper Roet, Ph.D., CEO and co-founder of QurAlis. “Loss of Kv7.2/7.3 function from the mis-splicing of the KCNQ2 gene leading to hyperexcitability-induced neurodegeneration was one of the first genetic breakthroughs from human ALS motor neuron stem cell models made by our founders at Harvard. QurAlis was started to develop a best-in-class Kv7 ion channel opener for the treatment of hyperexcitability-induced disease progression in ALS, which occurs in about half of all people living with ALS. Elevated SDTC has been clinically linked to faster disease progression and mortality, underscoring the need for an effective treatment for this population.”
Dr. Roet continued, “The decrease in SDTC observed with QRL-101 in this study was approximately 50% greater than previously reported for the Kv7 opener ezogabine in a single-dose study in people with ALS. QRL-101 has the potential to be a promising therapeutic, especially considering the encouraging results from a previous study with ezogabine in ALS patients showing positive effects on both SDTC and the disease progression biomarker compound muscle action potential (CMAP). Further, based on the strong clinical validation of Kv7.2/7.3 ion channel openers in seizure reduction, the TMS-EMG intracortical facilitation together with the EEG data from this study suggest QRL-101 may have the potential to reduce both frequency and severity of seizures in people living with epilepsy without causing sedation. We are encouraged by these results and look forward to advancing the clinical program for QRL-101 in both ALS and epilepsy so that we can fulfill our mission of bringing much-needed precision medicine options to patients.”
Kv7.2/7.3 is a voltage-gated potassium channel whose role is crucial for the regulation of both neuronal excitability and membrane potential. QRL-101 is a potentially best-in-class selective Kv7.2/7.3 ion channel opener for the treatment of hyperexcitability-induced disease progression in ALS, which occurs in both sporadic and genetic forms of ALS, with the majority caused by the mis-splicing of the KCNQ2 gene in the pre-mRNA. Kv7 is also a clinically validated target to regulate the hyperexcitable state in epilepsy. In vivo and in vitro studies have demonstrated that QRL-101 is more potent in threshold track studies and exhibits the potential for fewer clinical adverse events that ezogabine, a less selective, first-generation, Kv7.2/7.3 channel opener.
The Phase 1 PoM study (QRL-101-05, NCT06681441) was a randomized, double-blind, placebo-controlled, three-way crossover trial. The study was conducted at the Centre for Human Drug Research, an earlyphase Contract Research Organization based in Leiden, The Netherlands. The study evaluated two dose levels of QRL-101 versus placebo in healthy volunteers, assessing:
- Co-primary endpoints:
- Motor nerve excitability (mNETT SDTC; ALS biomarker)
- Cortical excitability (TMS-EMG MEP amplitude; epilepsy biomarker)
- Secondary endpoints:
- Additional TMS-EMG cortical excitability measures
- Resting-state pharmaco-EEG
- Safety, tolerability, and pharmacokinetics (PK)
Additional QRL-101 clinical trials include:
- QRL-101-01 (NCT05667779) was a first-in-human, single-ascending dose clinical trial in 88 healthy participants. In this clinical trial, QRL-101 was shown to be well tolerated. The study completed in late 2023 and results from QRL-101-01 supported a tolerable dose range for subsequent studies.
- QRL-101-03 (NCT06532396) is a randomized, double-blind, placebo-controlled, multiple-ascending dose clinical trial evaluating the safety, tolerability, and PK of QRL-101 in up to 60 healthy participants.
- QRL-101-04 (NCT06714396) is a Phase 1 PoM single-dose, placebo-controlled clinical trial designed to evaluate the safety and tolerability of QRL-101 in people living with ALS. QRL-101-04 is expected to enroll approximately 12 participants with ALS and will evaluate the impact of QRL-101 on mNETT.
- QRL-101-06 is a Phase 1 randomized, open-label, single dose, cross-over study to evaluate the PK of three formulations of QRL-101 in a fasted condition or in the presence of a high fat meal in healthy participants.
More information about the QRL-101 clinical trials can be found at www.clinicaltrials.gov.
About QurAlis Corporation
At QurAlis, we are neuro pioneers on a quest to cure, boldly seeking to translate scientific breakthroughs into powerful precision medicines. We work collaboratively with a relentless pursuit of knowledge, precise attention to craft, and compassion to discover and develop medicines that have the potential to transform the lives of people living with neurodegenerative and neurological diseases. QurAlis is the leader in development of precision therapies for amyotrophic lateral sclerosis (ALS). In addition to ALS, QurAlis is advancing a robust precision medicine pipeline to bring effective disease-modifying therapeutics to patients suffering from severe diseases defined by genetics and clinical biomarkers. For more information, please visit www.quralis.com or follow us on X @QurAlisCo or LinkedIn.
Contact:
Kathy Vincent
kathy.vincent@quralis.com
310-403-8951
- This investment will improve the healthcare experience for patients, reinforcing Dassault Systèmes’ role in the industry’s transformation
- Click Therapeutics will gain access to Dassault Systèmes’ global presence and expertise to support the development of new therapies
Dassault Systèmes (Euronext Paris: FR0014003TT8, DSY.PA) today announced its investment in Click Therapeutics, a leader in prescription digital therapeutics and software-enhanced drug therapies. The transaction advances Dassault Systèmes’ transformation of the patient experience in life sciences and healthcare through end-to end technology solutions used across the healthcare ecosystem.
The transaction strengthens the existing relationship between Dassault Systèmes’ MEDIDATA brand and Click Therapeutics, helping to improve patient engagement - post-trial through commercialization - and advancing the Patient Experience to deliver end-to-end technology solutions across the healthcare ecosystem. The continued relationship and subsequent investment will set a gold standard and unified offering for digital and pharmaceutical clinical trials design and execution, providing a path to Prescription Digital Therapeutics (PDTs) and SoftwareEnhanced (SE) product approvals, expanded labels for existing therapeutics, and, ultimately, a novel pipeline for long-term, real-world evidence generation.
MEDIDATA has virtualized clinical research, creating a unified patient experience with integrated technology solutions. This foundation, built on running over 8,000 active studies annually, allows MEDIDATA, together with Click Therapeutics, to extend their commitment to patients into realworld care, fostering improved coordination and outcomes between patients, physicians, caregivers, and life science manufacturers. Click Therapeutics will gain access to Dassault Systèmes’ global presence and expertise, while both companies will provide unparalleled support for developing PDTs and SE products, making them available to patients
“Digital therapeutics are beginning to transform the way customers think about their future clinical development programs and are providing demonstrable therapeutic benefits over drugs alone in many cases,” said Anthony Costello, CEO, Medidata. “Today’s investment in Click Therapeutics reflects our commitment to this exciting technology, and our partnership with them will enable our customers to take the fastest, most reliable path to bringing digital therapeutics to patients, helping to build a direct connection to patients over a life-long healthcare journey.”
“Since the FDA shared its latest thinking on Prescription Drug Use-Related Software in guidance, we have experienced a surge in interest in how software can add to the benefits of medication. Now is the time to expand treatment options for patients through software as medicine,” said David Benshoof Klein, CEO, Click Therapeutics. “The investment from Dassault Systèmes, the foremost leader in virtualizing the life sciences and healthcare industry, will enable us to continue to expand the boundaries of medicine and develop new, cutting-edge patient solutions to advance care.”
###
FOR MORE INFORMATION
Digital Therapeutics (DTx) are evidence-based software interventions designed to prevent, manage, or treat medical conditions, used independently or alongside traditional treatments. Prescription Digital Therapeutics (PDTs) are a subset of DTx that offer clinically validated treatment benefits. As such, they are regulated as medical devices, reviewed by regulatory authorities for safety and effectiveness, and require a healthcare provider’s prescription.
Software-enhanced (SE) drugs are drug-software combination products designed to enhance the value of a specific drug, built on the rigor and principles of PDTs. Software-enhanced drugs co-package a branded medication with a digital therapeutic that targets the unique needs of the medication and its patient population to deliver added clinical benefit compared to the medication alone. This creates an “SE” formulation of the drug that offers combined therapeutic benefits in one prescription for ease of use and optimal patient access.
About Medidata: Medidata is powering smarter treatments and healthier people through digital solutions to support clinical trials. Celebrating 25 years of ground-breaking technological innovation across more than 35,000 trials and 11 million patients, Medidata offers industry-leading expertise, analytics-powered insights, and the largest patient-level historical clinical trial data set in the world. More than 1 million registered users across approximately 2,300 customers trust Medidata’s seamless, end-to-end platform to improve patient experiences, accelerate clinical breakthroughs, and bring therapies to market faster. A Dassault Systèmes brand (Euronext Paris: FR0014003TT8, DSY.PA), Medidata is headquartered in New York City and has been recognized as a Leader by Everest Group and IDC. Discover more at www.medidata.com and follow us @Medidata.
About Click Therapeutics: Click Therapeutics, Inc. develops, validates, and commercializes software as prescription medical treatments for people with unmet medical needs. We are expanding the possibilities of medicine with Digital Therapeutics™ that combine clinical science with the power of software to create a new way to treat disease. Operating at the intersection of biology and technology, we use a proprietary platform-based approach to therapeutic development that leverages patient-centric design principles and innovative AI-based technologies to deliver a unique combination of engagement and clinical outcomes, consistently. Digital therapeutics on Click’s platform are regulated, clinically validated prescription mobile applications that can address diverse areas of therapeutic need, including indications in psychiatry, neurology, oncology, immunology, and cardiometabolic diseases. Click Therapeutics, in collaboration with Otsuka, developed the first prescription digital therapeutic authorized by the FDA for the adjunctive treatment of major depressive disorder symptoms, Rejoyn™. In October 2024, in response to FDA guidance on prescription drug use-related software (PDURS), we launched Click SE™ to extend our digital therapeutics platform and expertise to the development of software-enhanced drug™ therapies that combine software with pharmacotherapy to offer added clinically meaningful benefit to patients. Our commitment to advancing digital medicine means we continually improve our platform technologies, ensuring we stay at the forefront of cognitive, behavioral, and neuromodulatory therapeutic innovation, to achieve the best possible outcomes for patients. Our diverse team of innovators—spanning clinicians, researchers, technologists, designers and more—works together to create cutting-edge digital therapeutics, united in the mission to transform patient care. For more information, visit www.clicktherapeutics.com and connect with us on LinkedIn
About Dassault Systemes: Dassault Systèmes is a catalyst for human progress. Since 1981, the company has pioneered virtual worlds to improve real life for consumers, patients and citizens. With Dassault Systèmes’ 3DEXPERIENCE platform, 370,000 customers of all sizes, in all industries, can collaborate, imagine and create sustainable innovations that drive meaningful impact. For more information, visit: www.3ds.com
Dassault Systèmes Press Contacts
Corporate / France
Arnaud MALHERBE
arnaud.malherbe@3ds.com
+33 (0)1 61 62 87 73
North America
Natasha LEVANTI
natasha.levanti@3ds.com
+1 (508) 449 8097
EMEA
Virginie BLINDENBERG
virginie.blindenberg@3ds.com
+33 (0) 1 61 62 84 21
China
Grace MU
grace.mu@3ds.com
+86 10 6536 2288
Japan
Reina YAMAGUCHI
reina.yamaguchi@3ds.com
+81 90 9325 2545
Korea
Jeemin JEONG
jeemin.jeong@3ds.com
+82 2 3271 6653
India
Priyanka PANDEY
priyanka.pandey@3ds.com
+91 9886302179

- Financing led by funds managed by Blue Owl Capital; Senior Managing Director Kevin Raidy joins Latigo’s board
- Proceeds support advancement of company’s Nav1.8 inhibitor clinical programs and development of broader pipeline
THOUSAND OAKS, Calif. – March 17, 2025 – Latigo Biotherapeutics (“Latigo”), a clinical-stage biotechnology company developing best-in-class non-opioid pain treatments that target pain at its source, today announced it has closed $150 million in a Series B financing. Proceeds from the financing will support the advancement of the company’s highly selective Nav1.8 inhibitors currently in clinical development for the non-opioid treatment of pain, as well as the development of Latigo’s broader pipeline.
The financing was led by funds managed by Blue Owl Capital, with participation from Deep Track Capital, Access Biotechnology, Qatar Investment Authority, Cormorant Asset Management, Sanofi Ventures, Rock Springs Capital, UPMC Enterprises, and Kern Capital. Existing investors Westlake Village BioPartners, Foresite Capital, 5AM Ventures, and Alexandria Venture Investments also participated in the financing round, reaffirming their commitment to Latigo’s mission to develop safer, more effective pain treatments.
“The need for non-opioid pain treatments has never been more urgent, and this financing allows us to accelerate the development of our robust portfolio of pain medicines that have the potential to transform the treatment landscape,” said Nima Farzan, chief executive officer of Latigo Biotherapeutics. “We appreciate the support of our new and existing investors as we work to bring best-in-class, non-addictive pain treatments to patients.”
As part of the financing, Kevin Raidy, senior managing director at Blue Owl Capital, has joined Latigo’s board of directors. “The field of pain management is long overdue for innovation beyond opioids, and we believe Latigo is well-positioned to advance novel, non-addictive treatments that could make a real difference for patients,” Raidy said. “We are pleased to welcome Kevin to the board. His experience in life sciences investing and strategic growth will be invaluable as Latigo continues to scale,” said Timothy P. Walbert, chair of Latigo’s board. “With this financing and the continued support of world-class investors, we will be able to rapidly advance our pipeline of non-opioid treatments for chronic and acute pain.”
About Latigo Biotherapeutics’ Clinical Programs
Latigo recently reported positive Phase 1 results for LTG-001, its lead potential best-in-class non-opioid pain medicine candidate. LTG-001 is an oral, selective Nav1.8 inhibitor in development to treat acute pain at its source. In the Phase 1 first-in-human clinical trial, data showed that LTG-001 was well tolerated with rapid absorption.
LTG-305, oral, selective Nav1.8 inhibitor, currently in Phase 1 clinical trials, is a potential best-in- class non-opioid therapeutic candidate for the treatment of chronic pain. The Phase 1 trial is designed to evaluate the safety, tolerability, and pharmacokinetics of LTG-305 in healthy volunteers through single-ascending dose and multiple-ascending dose cohorts.
About Latigo Biotherapeutics
Latigo Biotherapeutics is a private clinical-stage biotechnology company developing innovative non-opioid pain medicines with potential best-in-class profiles that directly target the source of pain. Latigo’s goal is to provide effective, rapid-acting pain relief without the risk of addiction. For more information, please visit www.latigobio.com or follow us on LinkedIn.
Media Contact:
Kathy Vincent
Greig Communications Inc.
- Amezosvatein has shown excellent immunogenicity and improved tolerability in Phase 2 study
- Medicxi leads round, joined by new investors OrbiMed, HBM Healthcare Investments, and Sanofi Ventures
- Former Chair of GSK’s vaccine business and Chief Scientific Advisor to Operation Warp Speed, Moncef Slaoui, to join Curevo as Board Chair
Seattle, WA – March 17, 2025 – Curevo Vaccine (Curevo), a privately-held clinical-stage biotechnology company dedicated to developing varicella zoster virus (VZV) vaccines with improved tolerability, today announces the closing of a $110 million Series B round to advance development of amezosvatein, its vaccine for shingles.
Leading the round is new investor Medicxi, a European biotech-focused investment firm with significant experience investing in vaccine companies such as Vaxcyte and ViceBio. Joining Medicxi is an international group of investment funds including OrbiMed, HBM Healthcare Investments, and Sanofi Ventures plus existing investors RA Capital Management, Janus Henderson Investors, Adjuvant Capital, and founding investor GC Biopharma.
“This Series B round will fund the extension of our successful Phase 2 program into an additional 640 participants, including the key population of adults over age 70, to finalize dose selection ahead of the Phase 3 program,” said Curevo’s CEO, George Simeon, MBA/MPH.
“Designed based upon feedback from regulators and other stakeholders, this short extension trial will begin mid-2025 and serve to set the company for clinical, strategic, and regulatory success.”
Concurrent with the round, Moncef Slaoui, PhD, will join Curevo as Board Chair. Dr. Slaoui was most recently the Chief Scientific Advisor to Operation Warp Speed during the COVID-19 pandemic, helping deliver vaccines against the SARS-CoV-2 virus. He spent nearly 30 years at GlaxoSmithKline, during which he helped shape its vaccine business by contributing to the creation of numerous new vaccines including Shingrix® (shingles), Cervarix® (HPV-induced cervical cancer), Mosquirix® (malaria), Rotarix® (rotavirus gastroenteritis), and Synflorix® (pneumococcal disease).
“I have been collaborating with the Curevo team for several weeks now,” stated Dr. Slaoui. “I’m very excited to work with them to help perfect shingles vaccination, adding good tolerability to the exceptional efficacy achieved by the current vaccine. The data so far show Curevo’s adjuvant technology has the attributes to succeed in this endeavor.”
Also joining Curevo’s Board of Directors will be Giovanni Mariggi, PhD, co-founder and Partner at Medicxi, an experienced infectious disease investor and board member. “Patients, doctors, and payors are very clear a new shingles vaccine like amezosvatein would be welcome in the global marketplace. Amezosvatein’s activity and improved tolerability profile could allow it to be a significant product in the shingles vaccines market. We are excited to support Curevo’s plan with a streamlined path to approval,” stated Dr. Mariggi.
Tal Zaks, MD/PhD, will be joining Curevo’s Board of Directors on behalf of OrbiMed. Dr. Zaks is the former Chief Medical Officer of Moderna, where he led development of their mRNA vaccine against the SARS-CoV-2 virus. He previously held senior leadership positions in drug
development at GSK and Sanofi. “OrbiMed has a long history of investing in biotech companies like Curevo seeking to improve healthcare outcomes while creating long-term shareholder value,” said Dr. Zaks. “The fact amezosvatein contains an optimized version of the TLR4 agonist with a known mechanism of action reduces biological risk while providing the competitive advantage of potentially improved tolerability, which should position it as the first choice for people who want to be protected from shingles.”
About amezosvatein
‘Amezosvatein’ is the assigned non-proprietary name for CRV-101, a non-mRNA adjuvanted subunit vaccine under investigation by Curevo. Like Shingrix, amezosvatein uses a subunit protein antigen called glycoprotein ‘E’ (gE). Targeting the gE antigen is proven to elicit a long-term, protective immune response to prevent shingles. Amezosvatein’s adjuvant contains an optimized version of the TLR4 agonist proven by Shingrix to be biologically active in shingles vaccination. Amezosvatein was engineered to maintain exceptional efficacy and have a best-in-class tolerability profile. The SLA-SE adjuvant formulation was developed at Seattle-based Access to Advanced Health Institute (AAHI) and amezosvatein was licensed from the Mogam Institute for Biomedical Research, a research institute funded by South Korea’s GC Biopharma.
About Curevo
Curevo is a privately held, clinical-stage biotechnology company based near Seattle dedicated to reducing the burden of infectious disease by developing vaccines with improved tolerability and accessibility. Curevo’s lead product is amezosvatein, a non-mRNA adjuvanted sub-unit vaccine to prevent shingles, a serious medical condition involving a painful, blistering skin rash where 10-18% of people also develop serious, long-lasting nerve pain. The current $4+ billion shingles vaccine market is characterized by accessibility issues and vaccine hesitancy/dose avoidance related to vaccine tolerability. For more information visit https://curevovaccine.com/.
About Medicxi
Medicxi is a healthcare-focused investment firm with the mission to create and invest in companies across the full drug development continuum. Leveraging deep expertise in drug development and company creation spanning over two decades, Medicxi invests in early and late-stage therapeutics with a product vision that can fulfill a clear unmet medical need. For more information, please visit: https://www.medicxi.com.
About OrbiMed
OrbiMed is a leading global healthcare investment firm with approximately $17 billion in assets under management. OrbiMed invests globally across the healthcare industry, from start-ups to
large multinational corporations, via private equity funds, public equity funds, and royalty/credit funds. OrbiMed seeks to be a capital provider of choice, providing tailored financing solutions and extensive global team resources to help build world-class healthcare companies. OrbiMed’s team of over 100 professionals is based in New York City, London, San Francisco, Shanghai, Hong Kong, Mumbai, Herzliya, and other key global markets. For more information, please visit www.orbimed.com.
About HBM Healthcare Investments
Switzerland-based HBM Healthcare Investments focuses on the healthcare sector, holding and managing an international portfolio of promising companies in the human medicine, biotechnology, medical technology, diagnostics, and related areas. Many of these companies have their lead products already available on the market or are at an advanced stage of development. The portfolio companies are closely tracked and actively guided in their strategic direction. This is what makes HBM Healthcare Investments an interesting alternative to investments in big pharma and biotechnology companies. HBM Healthcare Investments has an international shareholder base and is listed on SIX Swiss Exchange (SIX:HBMN).
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi, focused on investing in promising early-stage healthcare companies. The firm supports pioneering innovations in biotechnology, digital health, and life sciences aligning with Sanofi’s mission to bring life-changing treatments to patients worldwide. For more information, please visit www.sanofiventures.com.
About RA Capital
Founded in 2004, RA Capital Management is a multi-stage investment manager dedicated to evidence-based investing in public and private healthcare, life sciences, and planetary health companies. RA Capital creates and funds innovative companies, from private seed rounds to public follow-on financings, allowing management teams to drive value creation from inception through commercialization and beyond. RA Capital's knowledge engine is guided by its TechAtlas internal research division, and Raven, RA Capital's healthcare incubator, offers entrepreneurs and innovators a collaborative and comprehensive platform to explore the novel and the re-imagined. RA Capital has more than 150 employees and over $10 billion in assets under management.
About Janus Henderson Investors
Janus Henderson Investors exists to help clients achieve their long-term financial goals. Its active management offers clients the opportunity to outperform passive portfolios over the course of market cycles. With more than 360 investment professionals, Janus Henderson Investors provides access to some of the industry’s most talented and innovative thinkers, spanning equities, fixed income, multi-asset, and alternatives, globally. Its investment teams blend insight, originality, and precision with rigorous analysis, structured processes, and robust risk management. Janus Henderson Investors builds client partnerships on openness and trust, channeling expertise from across the business and communicating the views of its experts in a timely and relevant way. https://www.janushenderson.com/
About Adjuvant Capital
Headquartered in New York, with offices in Zürich, Adjuvant is a global life science investment fund built to accelerate the development of new technologies for the world's most pressing public health challenges. Backed by prominent healthcare and emerging market investors, Adjuvant draws upon its network of scientists, public health experts, biopharmaceutical industry veterans, and development finance professionals to identify new investment opportunities. Adjuvant invests in companies developing promising new vaccines, therapeutics, diagnostics, and medical devices targeting high-burden infectious diseases, maternal and child health, antimicrobial resistance, and malnutrition, with a commitment to make these interventions accessible globally. For more information, visit http://www.adjuvantcapital.com
About GC Biopharma
Founding investor GC Biopharma (formerly known as Green Cross Corporation) is a biopharmaceutical company headquartered in Yongin, South Korea. The company has over half a century of experience in the development and manufacturing of plasma derivatives and vaccines and expanded its global presence with the successful USA market entry of ALYGLO™ (intravenous immunoglobulin G) in 2024. In line with its mission to meet the demands of future healthcare, GC Biopharma continues to drive innovation by leveraging its core R&D capabilities in engineering of proteins, mRNAs, and lipid nanoparticle (LNP) drug delivery platforms to develop therapeutics for the field of rare disease as well as I&I (Immunology & Inflammation). To learn more about the company, visit https://www.gcbiopharma.com/eng/
Shingrix®, Cervarix®, Mosquirix®, Rotarix®, and Synflorix® are registered trademarks of GlaxoSmithKline, PLC.
Contacts
David Miller
Sr. Director of Strategic Communications
Professor Allan Bradley to transition to Chief Scientific Officer, and will continue to drive the Company’s scientific direction
10 March 2025; Cambridge, England – T-Therapeutics, a biotechnology company developing next-generation soluble T cell receptor (TCR) therapeutics targeting cancer and autoimmune indications, announces the appointment of Theodora Harold as its new Chief Executive Officer (CEO). Founder and current CEO of T-Therapeutics Professor Allan Bradley will transition to the role of Chief Scientific Officer.
Theodora has over 25 years’ experience in the biotech sector, with a proven track record of dynamic leadership. She has raised significant capital for multiple companies as well as originating and driving numerous, high-profile, business development deals. She brings strong prior experience at the Board level and has held executive leadership positions in development and clinical stage oncology-focused companies, most recently as CEO of Crescendo Biologics. Theodora qualified as a Chartered Accountant with PricewaterhouseCoopers and graduated from the University of Cambridge.
Since its $60 million Series A fundraise at the end of 2023, T-Therapeutics has launched its OpTiMus® platform which is able to address previously intractable TCR targets with fully human, high affinity binders. The OptTiMus platform has now been used to generate a pipeline of first-in-class TCR therapeutics for cancer indications as well as inflammatory disorders. Theodora’s appointment as CEO is designed to support the Company’s transition into the development phase of its highly differentiated assets.
Professor Allan Bradley, Chief Scientific Officer of T-Therapeutics, said: “I am incredibly proud of what our team has already achieved - from developing a best-in-class transgenic mouse platform to securing the support of high-quality investors, and recently demonstrating ground-breaking preclinical activity with our T cell engaging molecules. Now is the right time to hand on the baton and I’m convinced that Theodora’s energetic leadership, capital markets expertise and deep understanding of the T cell landscape will take us further and faster towards our goal of bringing our next-generation TCR medicines to patients. As CSO, I look forward to working closely with her in the months and years ahead.”
Theodora Harold, newly-appointed Chief Executive Officer of T-Therapeutics, said: “I have been very impressed by T-Therapeutics’ world-leading science, the passion of the team and the first-in-class pipeline. The OpTiMus platform is uniquely able to address certain novel TCR targets and thus has the potential to transform the clinical landscape in both cancer and autoimmune diseases. I look forward to working with the Board of Directors, Allan and the whole team as we take the business into the next phase of its development.”
Dr David Hung, Chairman of T-Therapeutics, added: “Theodora’s appointment as our new CEO is a natural next step for T-Therapeutics and I’m delighted we have secured someone of such calibre to the team, who brings a wealth of complementary knowledge and experience. The team will continue to benefit from Allan’s expertise and leadership in his new role as CSO, overseeing the scientific development of our product candidates.”
Contact Us
T-Therapeutics
Theodora Harold
info@t-therapeutics.com
ICR Healthcare
Amber Fennell, Lucy Featherstone
t-therapeutics@icrhealthcare.com
About T-Therapeutics
T-Therapeutics is a next-generation T cell receptor (TCR) company spun out from the University of Cambridge. The company was created to harness the power of T cell biology, evolved over millions of years, to create safe and effective treatments for many cancers and autoimmune diseases. T-Therapeutics combines world-leading expertise in mouse genome engineering, deep knowledge and experience in biopharmaceutical drug development, single cell genomics, machine-learning and structural biology, anchored in a culture of creativity and collaboration. T-Therapeutics is developing ‘optimal’ TCR therapeutics using a proprietary OpTiMus® platform, based on a fully humanized TCR mouse that provides an almost unlimited source of unique, antigen-specific human TCRs. The company is developing a pipeline of first-in-class drugs that are intended to become transformative medicines, reshaping the clinical landscape for patients with cancer or autoimmune diseases.
The company is backed by blue-chip investors, including Sofinnova Partners, F-Prime, Digitalis Ventures, Cambridge Innovation Capital and Sanofi Ventures.
Oxford, United Kingdom – 26 February 2025 – OMass Therapeutics (‘OMass’ or ‘the Company’), a biotechnology company identifying medicines against highly validated target ecosystems such as membrane proteins or intracellular complexes, today announces that Birgitte Volck MD, PhD has joined its Board as a non-executive director.
Dr Volck has over 25 years of industry experience spanning R&D leadership, platform innovation, and commercialization, with a strong focus on rare diseases and precision medicine. She currently serves on the Board of Soleno Therapeutics and has held Board roles at many leading biotech companies including Nykode Therapeutics, Wilson Therapeutics, and Ascendis Pharma.
Her prior executive roles include Executive Vice President and Interim Chief Medical Officer at Ascendis Pharma, where she led clinical development, regulatory affairs, and medical strategy for the company’s rare disease portfolio and President of R&D at Avrobio Inc, where she oversaw the development of the company’s gene therapy programs. She also served as Senior Vice President, Head of R&D, Rare Diseases at GSK, where she helped shape the company’s rare disease portfolio leveraging internal and external partnerships. Other prior executive leadership roles include developing and launching innovative therapies for specialty care and rare disease indications at Sobi, Amgen, and Genzyme. Furthermore, she has contributed to the rare disease and precision medicine field as speaker and external lecturer. Birgitte earned her MD and PhD in Biomarkers for Arthritis from Copenhagen University.
Dr Volck, non-executive Director at OMass Therapeutics commented, “I have been following the OMass story for a number of years and have been impressed by its innovative approach to drug discovery and the study of protein interactions in their native ecosystem, as well as the Company’s leadership and strategic focus. I am passionate about developing new medicines and look forward to using my experience and network to help guide the team as they prepare to enter the clinic with their potentially best-in-class MC2 antagonist for congenital adrenal hyperplasia and ACTHdependent Cushing’s.”
Jim Geraghty, Chairman of OMass Therapeutics’ Board of directors added, “Birgitte is very well known in the rare diseases community and has a wealth of experience in developing drugs in numerous indications. Ros, the Board and I look forward to capitalising on this deep experience as we approach the next step in our journey as a clinical stage company.”
OMass Therapeutics
Rosamond Deegan, Chief Executive Officer
Phone: +44 (0) 1235 527589
Email: ros.deegan@omass.com
ICR Healthcare
Sue Charles / Ashley Tapp
Phone: +44 (0)20 3709 5700
Email: omass@icrhealthcare.com
About OMass Therapeutics
OMass Therapeutics is a biotechnology company discovering medicines against highly-validated target ecosystems, such as membrane proteins or intracellular complexes.
OdyssION™, OMass’ unique drug discovery platform, comprises next-generation native mass spectrometry with novel biochemistry techniques and custom chemistry to interrogate not just a drug target, but also the interaction of the target with its native ecosystem, separate from the confounding complexity of the cell. This unique approach results in cell-system fidelity with cell-free precision.
OMass is advancing a pipeline of small molecule therapeutics in rare diseases and immunological conditions. Its lead programme is a best-in-class MC2 (melanocortin-2) receptor antagonist for the treatment of Congenital Adrenal Hyperplasia (CAH) and Cushing’s Syndrome. The focus of the program has been to increase receptor residency time to make OMass’ antagonists resistant to competition by the endogenous ligand, thereby avoiding loss of efficacy in the face of rising adrenocorticotropic hormone (ACTH) levels due to reductions in glucocorticoid supplementation for CAH or progression of Cushing’s Syndrome.
Headquartered in Oxford, UK, OMass has raised over $160M (£129M) from a top-tier international investor syndicate including Syncona, Oxford Science Enterprises, GV, Northpond Ventures, Sanofi Ventures and British Patient Capital.
To learn more, please visit www.omass.com. Follow us on LinkedIn.
CAMBRIDGE, Mass., February 24, 2025 – QurAlis Corporation (“QurAlis”), a clinical-stage biotechnology company driving scientific breakthroughs into powerful precision medicines that have the potential to alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative and neurological diseases, today announced that company management will participate in the following investor conferences in March.
T.D. Cowen 45th Annual Health Care Conference (March 3-5, 2025)
Format: One-on-one investor meetings
Location: Boston, MA
Stifel 2025 Virtual CNS Forum (March 18-19, 2025)
Format: Corporate presentation
Date: Wednesday, March 19, 2025
Time: 3:30 PM ET
Location: Virtual
The QurAlis corporate presentation can be accessed by visiting the presentations section of the Company’s website at www.quralis.com.
About QurAlis Corporation
At QurAlis, we are neuro pioneers on a quest to cure. We work with a relentless pursuit of knowledge, a precise attention to craft, and an optimistic mindset to discover and develop effective precision medicines that have the potential to alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative and neurological diseases. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a robust precision medicine pipeline with therapeutic candidates aimed at modifying severe disease pathology in defined patient populations based on both disease-causing genetic mutation(s) and clinical biomarkers. For more information, please visit www.quralis.com or follow us on X @QurAlisCo or LinkedIn.
Contact: Kathy Vincent kathy.vincent@quralis.com 310-403-8951

- Series C financing led by NEA, joined by new investor Foresite Capital and Abcuro’s existing investors
- Proceeds to support completion of the registrational Phase 2/3 MUSCLE trial of ulviprubart in inclusion body myositis, BLA filing, and commercial launch preparation
Newton, Massachusetts, February 12, 2025— Abcuro, Inc., a clinical-stage biotechnology company developing therapies for the treatment of autoimmune diseases and cancer through precise modulation of cytotoxic T cells, today announced the closing of a $200 million Series C financing led by New Enterprise Associates (NEA) with Foresite Capital joining the round and participation of existing investors including RA Capital Management, Bain Capital Life Sciences, Redmile Group, Samsara BioCapital, Sanofi Ventures, Pontifax, Mass General Brigham Ventures, New Leaf Ventures, funds managed by abrdn Inc., funds and accounts managed by BlackRock, Eurofarma Ventures, and Soleus Capital.
Proceeds from the Series C financing will be used to complete the registrational Phase 2/3 MUSCLE clinical trial evaluating ulviprubart (ABC008), a first-in-class monoclonal antibody targeting killer cell lectin like receptor G1 (KLRG1), for the treatment of inclusion body myositis (IBM). Assuming positive results from the MUSCLE clinical trial, Abcuro plans to file a BLA and will use a portion of the proceeds to support commercial launch preparation.
“Continued support from all of our investors in this latest financing round validates our vision for the potential that ulviprubart may have as a novel treatment for progressive and devastating diseases mediated by highly cytotoxic T cells, including Inclusion Body Myositis” said Alex Martin, Chief Executive Officer of Abcuro. “We are in a strong position to execute on our clinical development plan, including completing our ongoing, registrational Phase 2/3 MUSCLE clinical trial of ulviprubart in IBM, and expect to report initial data in the first half of 2026. We will also look to fund the expansion of manufacturing capabilities and other pre-commercial activities this year.”
“Abcuro represents an exciting opportunity with its lead candidate, ulviprubart, a potential first-in-class therapy that could make a big impact to the treatment paradigm of IBM, an indication with a significant unmet clinical need,” said Michele Park, PhD, Partner at NEA. “Ulviprubart targets a unique mechanism that can selectively deplete cytotoxic T cells, backed by encouraging clinical and preclinical data that have been presented to date.”
About Ulviprubart
Ulviprubart (ABC008) is a first-in-class anti-KLRG1 antibody product candidate capable of selectively depleting highly cytotoxic T cells, while sparing naïve, regulatory and central memory T cells. Ulviprubart is designed to treat diseases mediated by highly cytotoxic T cells, including the autoimmune muscle disease inclusion body myositis (IBM) and T cell large granular lymphocytic leukemia (T-LGLL). The US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have each granted orphan drug designation to ulviprubart for the treatment of IBM.
About Inclusion Body Myositis (IBM)
IBM is an autoimmune disease in which highly cytotoxic T cells chronically attack muscle tissue leading to progressive weakness and limb muscle atrophy. Patients progressively lose muscle function, including loss of grip, dexterity and mobility. There are currently no available disease-modifying treatment options for IBM. Based on published epidemiology literature, it is estimated that there are more than 50,000 people with IBM across the US and Europe.
About Abcuro
Abcuro is a clinical stage biotechnology company developing first-in-class immunotherapies for the treatment of autoimmune diseases and cancer through precise modulation of highly cytotoxic T cells. The company’s lead program is ulviprubart (ABC008) and is currently in clinical trials for inclusion body myositis (IBM) and T cell large granular lymphocytic leukemia.
For more information, visit us on LinkedIn and at abcuro.com.
Contact:
Matthew DeYoung
Investor Relations and Media
Argot Partners
abcuro@argotpartners.com
Brisbane, Australia and Indianapolis, USA – February 10, 2025 – AdvanCell, a clinical-stage radiopharmaceutical company specializing in targeted alpha therapies, today announced an expansion to the scope and breadth of its strategic collaboration with Eli Lilly and Company to research and develop innovative treatments for various cancers.
Under this new agreement, the parties will leverage AdvanCell’s proprietary Pb-212 production technology and radionuclide development infrastructure and Lilly’s drug candidate programs and extensive expertise in drug development to facilitate the development and accelerate the clinical advancement of an expanded portfolio of targeted alpha therapies.
AdvanCell’s competitive advantage in technology development and the infrastructure it has built to accelerate early-stage clinical trials in Australia enables AdvanCell to rapidly develop and progress novel Pb-212-containing radiotherapeutics from discovery into clinical trials.
“This collaboration with Lilly represents a significant milestone for AdvanCell, recognizing our company as one of the leaders in the Pb-212 targeted alpha therapy space,” said Andrew Adamovich, CEO of AdvanCell. “By combining our groundbreaking isotope production capabilities, our team’s expertise and infrastructure with Lilly’s pharmaceutical and oncology expertise and global scale, we aim to bring transformative treatments to patients with hard-to-treat cancers. It is especially pleasing to continue and expand our existing relationship.”
Jacob Van Naarden, President of Lilly Oncology, added, “Partnering with AdvanCell aligns with our commitment to advancing innovative radiopharmaceuticals. We are excited to explore the potential of Pb-212-based alpha therapies as we work to bring meaningful new treatments for patients.”
Financial terms of the agreement were not disclosed.
About AdvanCell
AdvanCell is dedicated to developing innovative cancer therapies that harness the power of targeted alpha-emitting radionuclides. By combining advanced manufacturing capabilities with cutting-edge science and clinical development capabilities, AdvanCell aims to deliver novel treatments that improve outcomes for cancer patients globally. For more information, visit www.advancell.com.au.
Patient-Centered PRIZM Study Measures Impact of Once-Daily Oral Zagociguat on Fatigue and Cognitive Impairment, Hallmark Features of this Rare Mitochondrial Disease
CAMBRIDGE, Mass., January 27, 2025 – Tisento Therapeutics today announced that the first patient has been dosed in its global Phase 2b PRIZM study. The study is investigating the impact of once-daily oral
zagociguat treatment on fatigue, cognitive impairment, and other key aspects of the rare mitochondrial disease MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-like Episodes).
The PRIZM study is centered in multiple ways on what matters to people living with MELAS. The study incorporates features that maximize convenience for participants, including at-home assessments, the potential for at-home study visits, and door-to-door travel support. In addition, all participants will receive zagociguat during one period of this crossover study, and participants who complete the study may be eligible for continued access via an open-label extension study. Importantly, the clinical outcome
assessments and endpoint strategy for the PRIZM study were informed by Tisento’s interview study in which individuals living with MELAS described the symptoms and impacts of the disease that are most important to them. PRIZM is a global study now enrolling in North America, Europe, and Australia. Patients and families interested in learning more can visit Tisento’s PRIZM page, ClinicalTrials.gov (NCT06402123), or discuss with their physician.
“Since Tisento’s founding, we have been focused on initiating a thoughtful, well-designed clinical study to evaluate zagociguat for the treatment of MELAS, and we are humbled by the enthusiastic response we have received from physicians and the mitochondrial disease community,” said Peter Hecht, PhD, chief executive officer of Tisento. “We are pleased that the first participants are enrolling in the global PRIZM study, which was designed with patient perspectives at the forefront. We will continue to put patients first every step of the way as we work to develop creative solutions to address what matters most for people with serious diseases.”
“MELAS is the most common condition we see in our mitochondrial clinic, with profound effects on patients' daily lives and wellbeing. Right now, we have no approved treatments, making the search for effective therapies absolutely critical,” said Dr. Austin Larson, associate professor of pediatric clinical genetics and metabolism at the University of Colorado School of Medicine and PRIZM study investigator. “I am excited about zagociguat's potential to address both the severe fatigue and cognitive challenges that so significantly impact our MELAS patients.”
About the PRIZM Study
PRIZM – a Phase 2b Randomized, Placebo-Controlled Trial Investigating Zagociguat in MELAS – is evaluating the efficacy and safety of oral zagociguat 15 mg or 30 mg compared to placebo when administered once-daily for 12 weeks in participants with genetically and phenotypically defined MELAS. The PRIZM study has a crossover design, with two 12-week treatment periods separated by a 4-week washout period. All participants will receive zagociguat during one of the 12-week periods and placebo during the other. Participants who complete the study may be eligible for an open-label extension
study.
PRIZM is a global study that will enroll approximately 44 participants at mitochondrial disease centers of excellence in the U.S., Italy, Germany, United Kingdom, Australia, and Canada. For more information, please visit www.tisentotx.com/prizm or ClinicalTrials.gov (NCT06402123) for more information. Interested individuals can also reach out to their physicians for participation details.
About Zagociguat
Zagociguat is a once-daily, oral, clinical-stage investigational medicine with potential to positively impact both peripheral and central nervous system manifestations of mitochondrial diseases. Zagociguat stimulates soluble guanylate cyclase (sGC), an enzyme that is found in virtually every cell in every tissue of the body and is part of a system of cellular mechanisms that control critical physiological functions including neuronal function and blood flow.
A first-in-class, brain-penetrant sGC stimulator, zagociguat is hypothesized to rebalance dysregulated cellular pathways in MELAS. By restoring cellular functions that support mitochondria, zagociguat may help restore mitochondrial energy production and physiological function.
In a Phase 2a study in patients with MELAS, zagociguat exhibited a favorable safety profile, exposure throughout the body including in the central nervous system, and improvements in neuronal function, mitochondrial function, and blood flow in the brain. Zagociguat is currently being evaluated as a treatment for MELAS in the Phase 2b PRIZM study.
For more information, visit www.tisentotx.com/our-science.
About Tisento Therapeutics
Tisento Therapeutics, a privately held biotech company, is developing novel medicines to treat diseases with significant unmet need, beginning with MELAS and other genetic mitochondrial diseases. Tisento means “I hear you” in Italian; our approach to innovation begins with listening to patients and then channeling what we learn into decisive actions that shape our research and clinical programs.
Tisento is guided by a high-caliber internal team of biopharma veterans and an extensive external network of expert physicians, patient advocacy groups, researchers, industry-leading vendors, and other close collaborators who are partners in our mission to develop meaningful treatments for mitochondrial diseases.
Learn more at our website, www.tisentotx.com, or connect with us on LinkedIn (Tisento Therapeutics) or X (@tisentotx).
Contact
Tisento Media Relations
Jessi Rennekamp, Astrior Communications
Email: jessi@astriorcomms.com
Sydney, Australia – 3 February 2025 – AdvanCell, a clinical-stage radiopharmaceutical company developing innovative cancer therapeutics, today announced the successful completion of an oversubscribed US$112 million Series C financing. This milestone funding round was co-led by SV Health Investors, Sanofi Ventures, Abingworth, and SymBiosis. Additional support came from existing investor Morningside, alongside new investors Tenmile, Brandon Capital, and others.
Since its founding in June 2019, AdvanCell has grown from a belief in the potential of Targeted Alpha Therapy into a global organisation with 60 passionate team members, a 40,000-square- foot manufacturing facility, world-class pre-clinical infrastructure, a potentially best-in-class drug for prostate cancer that is in trials, and a deep and developing pipeline of assets.
This investment will fuel AdvanCell’s ongoing efforts to expand its manufacturing capacity, accelerate the clinical development of its pipeline of radionuclide therapies, and advance its mission of delivering life-changing treatments to cancer patients worldwide.
AdvanCell is currently enrolling patients for the highest dose cohort of its multi-center TheraPb Ph I/II dose escalation clinical trial of ADVC001 for metastatic prostate cancer, a potentially best-in-class Targeted Alpha Therapy. The trial aims to demonstrate the safety and efficacy of Pb-212-based radionuclide treatment.
As part of the financing, Jamil M. Beg from SV Health Investors, Christopher Gagliardi from Sanofi Ventures, and Bali Muralidhar from Abingworth have joined the AdvanCell Board of Directors. They bring extensive industry expertise to support the company’s growth and join existing directors Bill Ferris AC, Anthony Aiudi, Kevin Cameron, and Andrew Adamovich.
Jamil M. Beg, Partner at SV Health Investors, commented:
“We are delighted to support AdvanCell’s growth and play a role in its remarkable journey through leading this oversubscribed Series C. We have looked long and deep at the field of radionuclide therapies and are confident that AdvanCell stands out with a first-in-class Pb-212- PSMA program and a best-in-class manufacturing platform. The company’s exceptional team, technologies, robust infrastructure, collaborations with some of the world’s largest pharma companies and ability to consistently execute, position it to truly change outcomes for patients. We are excited to help AdvanCell realize its potential in transforming cancer care globally.”
AdvanCell was founded on the belief that Targeted Alpha Therapies could change the course of cancer treatment and that the scalable supply of isotopes would enable the development of multiple practice-changing drugs. Radionuclide therapies for prostate cancer and gastroenteropancreatic neuroendocrine tumours have transformed patient care. Pb-212 Targeted Alpha Therapy has the potential to further advance this progress by leveraging the radiobiological and physical attributes of Pb-212 to make life-changing treatments for patients.
“This successful Series C round demonstrates strong confidence in our vision and capabilities,” said Andrew Adamovich, CEO of AdvanCell. “We are grateful for the continued support from our existing investors, particularly the long-term support from Morningside and are excited to welcome new partners who share our commitment to transforming cancer care. With this funding, AdvanCell is well-positioned to scale our manufacturing operations and progress our cutting-edge therapies towards commercialization.”
The Series C funding represents a significant step in AdvanCell’s journey to become a global leader in radionuclide-based cancer therapeutics.
For media inquiries, please contact:
Andrew Adamovich CEO
Email: contact@advancell.com.au
Phone: +612 8000 4199
About AdvanCell
AdvanCell is dedicated to developing innovative cancer therapies that harness the power of targeted alpha-emitting radionuclides. By combining advanced manufacturing capabilities with cutting-edge science and clinical development capabilities, AdvanCell aims to deliver novel treatments that improve outcomes for cancer patients globally. For more information, visit www.advancell.com.au.
About SV Health Investors
SV Health Investors is a leading healthcare fund manager committed to investing in tomorrow’s healthcare breakthroughs. The SV funds invest across stages, geographic regions, and sectors, with expertise spanning biotechnology, dementia, medical devices, healthcare growth and healthcare technology. With approximately $2bn in assets under management and a truly transatlantic presence with offices in Boston and London, SV has built an extensive network of talented investment professionals and experienced industry veterans. Since its founding in 1993, SV has invested in, created and built more than 200 companies attracting global talent, entrepreneurs and pharma partners. To date, these investments have resulted in the licensing of 26 novel drugs and six new drug classes able to treat indications with unmet medical needs and deliver positive impact to patients. For more information, please visit www.svhealthinvestors.com.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi, focused on investing in promising early-stage healthcare companies. The firm supports pioneering innovations in biotechnology, digital health, and life sciences that align with Sanofi’s mission to bring life- changing treatments to patients worldwide. For more information, please visit www.sanofiventures.com.
About Abingworth
Abingworth is a leading transatlantic life sciences investment firm and part of the global investment firm Carlyle (NASDAQ: CG). Abingworth helps transform cutting-edge science into novel medicines by providing capital and expertise to top calibre management teams building world-class companies. Since 1973, Abingworth has invested in over 185 life science companies, leading to 50+ M&As and more than 75 IPOs. Our therapeutic focused investments fall into three categories: seed and early-stage, development stage, and clinical co- development. Abingworth supports its portfolio companies with a team of experienced professionals at offices in London, Menlo Park (California), and Boston. For more information, please visit www.abingworth.com.
About SymBiosis Capital Management
SymBiosis is an investment firm focused on advancing biotherapeutics innovations for serious and life-threatening diseases. The firm invests in groundbreaking medicines across disease areas, financing stages, and geography, with a focus on programs in, or about to enter, human trials. SymBiosis currently manages a portfolio of more than 30 investments and has significant, long-term capital commitments to fund future investments. For more information, please visit www.symbiosis.vc or follow us on LinkedIn.
– IND submissions planned in 2025 for first two programs within leading portfolio of di- siRNA therapies –
– Series B funds will support progressing programs for KCNT1-related epilepsy and Huntington’s disease through clinical proof-of-concept –
BOSTON, January 28, 2025 – Atalanta Therapeutics, a biotechnology company pioneering RNA interference (RNAi) for the treatment of neurological diseases, today announced the completion of a $97 million Series B financing to support Phase 1 clinical trials of the company’s investigational RNAi therapies for KCNT1-related epilepsy and Huntington’s disease. The financing was co-led by EQT Life Sciences and Sanofi Ventures, with participation from other new investors RiverVest Venture Partners, funds managed by abrdn Inc, Novartis Venture Fund, Pictet Alternative Advisors, Mirae Asset Financial Group, and GHR Foundation alongside existing investor F-Prime Capital.
“This financing validates the truly transformative potential of Atalanta’s best-in-class di-siRNA platform for delivering oligonucleotide therapies to the central nervous system and the exciting promise of our expansive wholly-owned pipeline,” said Alicia Secor, Atalanta’s president and chief executive oXicer. “Importantly, this Series B will support a path to the clinic for two programs for serious neurological diseases that today lack disease-modifying therapies — KCNT1-related epilepsy and Huntington’s disease — and will anchor our growing franchise of investigational medicines for Huntington’s disease. We are diligently progressing these programs toward IND submissions this year so that we can start our Phase 1 trials and reach patients who are waiting.”
In conjunction with this financing, the company announced the appointments of Arno de Wilde, M.D., Ph.D., MBA, managing director at EQT Life Sciences; Jason Hafler, Ph.D., managing director of Sanofi Ventures; and Niall O’Donnell, Ph.D., managing director of RiverVest Venture Partners to its Board of Directors.
“Atalanta’s di-siRNA technology has shown promising ability to durably silence disease-promoting genes throughout previously inaccessible regions of the brain and spinal cord — opening a wide range of treatment possibilities for devastating neurological diseases,” said Dr. de Wilde. “EQT Life Sciences is proud to co-lead this investment in Atalanta’s future as part of such a high-quality investor syndicate, and we look forward to partnering with Atalanta’s leadership to support their continued success.”
“We are excited to partner with Atalanta as they enter their next chapter as a clinical-stage company,” said Dr. Hafler. “Their success to-date is a strong validation of their ability to create meaningful new RNAi therapies, and Sanofi Ventures is glad to support Atalanta as they advance their pipeline.”
ATL-201 is Atalanta’s investigational therapy for KCNT1-related epilepsy, an early-onset seizure disorder and encephalopathy driven by gain-of-function variants in the KCNT1 gene. Infants and children with KCNT1-related epilepsy have severe, frequent seizures that are unable to be controlled with anti-seizure medications, and they often experience developmental delays and intellectual disability. ATL-201 is designed to reduce KCNT1 levels and to normalize neuronal excitability. Preclinical studies have shown that ATL-201 produces a significant reduction of seizures and improvement in behavior with impressive durability and tolerability.
The company’s second development candidate, ATL-101, is a di-siRNA designed to silence the HTT gene for the treatment of Huntington’s disease. Huntington’s disease is a progressive neurodegenerative disease caused by an expansion of the HTT gene, which leads to deterioration in a person’s physical, cognitive, and psychiatric abilities. Preclinical studies have shown that a single dose of ATL-101 produces a potent and strong reduction in HTT expression, including in deep brain regions, with six months of durability and excellent tolerability.
A full overview of the company’s pipeline, disclosed today, is available here.
About Atalanta Therapeutics
Atalanta Therapeutics is a biotechnology company developing treatments for intractable diseases of the central nervous system using RNA interference. Atalanta’s unique platform of divalent small interfering RNA (di-siRNA) enables durable, selective gene silencing throughout the brain and spinal cord. Atalanta is advancing a wholly owned pipeline of disease-modifying programs for Huntington’s disease, genetic epilepsy, severe chronic pain, and other neurological diseases in addition to partnered programs as part of a strategic collaboration with Genentech. Atalanta is headquartered in Boston, Mass. For more information, visit www.atalantatx.com.
Media Contact:
Michael Galfetti
Ten Bridge Communications
mgalfetti@tenbridgecommunications.com
CAMBRIDGE, Mass., January 7, 2025 – QurAlis Corporation (“QurAlis”), a clinical-stage biotechnology company driving scientific breakthroughs into powerful precision medicines that have the potential to alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative and neurological diseases, today announced that it has been invited to present at the 43rd Annual J.P. Morgan Healthcare Conference to be held January 13-16, 2025, in San Francisco, CA.
Kasper Roet, Ph.D., QurAlis’ chief executive officer and co-founder, will present a corporate overview on Monday, January 13, 2025 at 5:30PM PT at the Westin St. Francis in the Golden Gate room.
The QurAlis corporate presentation can be accessed by visiting the presentations section of the Company’s website at www.quralis.com.
About QurAlis Corporation
At QurAlis, we are neuro pioneers on a quest to cure. We work with a relentless pursuit of knowledge, a precise attention to craft, and an optimistic mindset to discover and develop effective precision medicines that have the potential to alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative and neurological diseases. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a robust precision medicine pipeline with therapeutic candidates aimed at modifying severe disease pathology in defined patient populations based on both disease-causing genetic mutation(s) and clinical biomarkers. For more information, please visit www.quralis.com or follow us on
X @QurAlisCo or LinkedIn.
Contact:
Kathy Vincent
kathy.vincent@quralis.com
310-403-8951
Financing co-led by Samsara Biocapital and Enavate Sciences, with participation from Pfizer Ventures and Regeneron Ventures in addition to other new and existing investors
Normunity’s lead program is a novel T cell engager against a tumor-specific target that is relevant in multiple solid tumors and has the potential to be leveraged for a range of biologic modalities
Boston, Mass., and West Haven, Conn. – January 13, 2024 – Normunity, a biotechnology company creating novel anti-cancer therapies, today announced that it has closed a Series B financing for $75 million. The financing was co-led by Samsara BioCapital and Enavate Sciences, alongside other new investors Regeneron Ventures, Pfizer Ventures and YK Bioventures, as well as existing investors Canaan Partners, Sanofi Ventures, Taiho Ventures, Osage Venture Partners, HongShan Capital Group, and Connecticut Innovations. The Board of Directors will be expanded to include David Parry, PhD (Samsara), Sara Nayeem, MD (Enavate) and Marie-Clare Peakman, MD (Pfizer).
“We are delighted to secure this financing from such an experienced and well-respected syndicate of life sciences investors. Their support will enable Normunity to continue our strong momentum and advance our lead drug program NRM-823 into the clinic this year and explore other modalities with this exciting tumor specific target,” said Rachel Humphrey, MD, founding Chief Executive Officer of Normunity. “With our proprietary target discovery process, we will continue to build our pipeline around novel targets, with new biological insights that could translate into life-changing medicines for cancer patients.”
Proceeds from the financing will be used to advance Normunity’s lead program, NRM‑823, a first-in-class T cell engager that binds a novel, highly specific tumor target expressed on multiple types of solid tumors. The company plans to initiate the Phase 1 clinical trial of NRM-823 in 2H 2025 and leverage its prior work by advancing other modalities against this tumor-specific target, including antibody‑drug conjugates and radiotheranostics. The funding will also be used to advance Normunity’s pipeline of programs that address novel targets responsible for tumor-specific immune suppression. These pipeline candidates derive from Normunity’s proprietary target discovery process, conducted in collaboration with the lab of Professor Lieping Chen, MD PhD, at the Yale University School of Medicine, and built on insights into untapped biological mechanisms that occur from the complex interactions of the immune system and cancer.
“Normunity has an outstanding team that has made impressive progress in discovering and advancing an exciting cancer drug program in NRM-823, which has the potential to address a previously unrecognized target that plays a role in supporting cancer survival across a range of solid tumors,” said David Parry, PhD, Venture Partner at Samsara BioCapital. “We look forward to working with the Normunity team as they continue to advance NRM-823 and other novel agents in the pipeline.”
“The T cell engager space has seen multiple recent successes in solid tumors, and we believe this modality is poised to transform the treatment of numerous cancers in the coming years,” said Sara Nayeem, MD, EVP, Investments at Enavate Sciences. “We are excited to support the development of NRM-823, which has many characteristics of an ideal T cell engager and has demonstrated compelling preclinical efficacy and safety.”
About Normunity
Normunity, is a biotechnology company creating novel anti-cancer therapies that address untapped biology at the interface of the immune system and the tumor to target mechanisms that impact tumor growth and circumvent immune surveillance and tumor clearance. The company is using these novel targets to build a pipeline of anti-cancer medicines, including therapeutic antibodies, bispecific antibodies, and payload-carrying biologics. The company’s lead program, NRM‑823, is a T cell engager with tumor-specific targeting for multiple solid tumors and is expected to enter the clinic in 2025. Normunity is located in Boston, MA, and New Haven, CT. For more information, please visit www.normunity.com and follow us on LinkedIn.
Media Contact
Kathryn Morris, The Yates Network LLC
914-204-6412

-
Acquisition adds potential best-in-class disease-modifying therapy for Alzheimer's disease, ALIA-1758, and novel blood-brain barrier (BBB)-crossing technology to strengthen neuroscience pipeline and R&D capabilities
NORTH CHICAGO, Ill., Dec. 11, 2024 /PRNewswire/ -- AbbVie (NYSE: ABBV) announced today that it has completed its acquisition of Aliada Therapeutics. With the completion of the acquisition, Aliada is now a part of AbbVie.
Aliada's lead investigational asset is ALIA-1758, an anti-pyroglutamate amyloid beta (3pE-Aβ) antibody, which is in development for the treatment of patients with Alzheimer's disease and is currently in a Phase 1 clinical trial. ALIA-1758 utilizes a novel blood-brain barrier (BBB)-crossing technology that enhances delivery of targeted drugs into the central nervous system (CNS).
"Alzheimer's disease poses a significant public health challenge, impacting millions worldwide and is becoming more prevalent as populations age," said Dawn Carlson, M.D., M.P.H., vice president, neuroscience development at AbbVie. "With the acquisition now complete, we look forward to advancing potentially disease-modifying therapies such as ALIA-1758 for Alzheimer's disease and bolstering our neuroscience discovery and development efforts by leveraging Aliada's novel CNS drug delivery platform."
For additional background on the acquisition, please read the announcement press release here.
About AbbVie
AbbVie's mission is to discover and deliver innovative medicines and solutions that solve serious health issues today and address the medical challenges of tomorrow. We strive to have a remarkable impact on people's lives across several key therapeutic areas – immunology, oncology, neuroscience, and eye care – and products and services in our Allergan Aesthetics portfolio. For more information about AbbVie, please visit us at www.abbvie.com. Follow @abbvie on LinkedIn, Facebook, Instagram, X (formerly Twitter), and YouTube.
Forward-Looking Statements
Some statements in this news release are, or may be considered, forward-looking statements for purposes of the Private Securities Litigation Reform Act of 1995. The words "believe," "expect," "anticipate," "project" and similar expressions and uses of future or conditional verbs, generally identify forward-looking statements. AbbVie cautions that these forward-looking statements are subject to risks and uncertainties that may cause actual results to differ materially from those expressed or implied in the forward-looking statements. Such risks and uncertainties include, but are not limited to, challenges to intellectual property, competition from other products, difficulties inherent in the research and development process, adverse litigation or government action, and changes to laws and regulations applicable to our industry. Additional information about the economic, competitive, governmental, technological and other factors that may affect AbbVie's operations is set forth in Item 1A, "Risk Factors," of AbbVie's 2023 Annual Report on Form 10-K, which has been filed with the Securities and Exchange Commission, as updated by its subsequent Quarterly Reports on Form 10-Q. AbbVie undertakes no obligation, and specifically declines, to release publicly any revisions to forward-looking statements as a result of subsequent events or developments, except as required by law.
For further information: Media: Elizabeth Tang, Ph.D., liz.tang@abbvie.com; Investors: Liz Shea, liz.shea@abbvie.com

Menlo Park, Calif., December 5, 2024 – Nuvig Therapeutics, Inc., (“Nuvig”) a privately held biotechnology company developing novel immunomodulatory therapeutics for patients with inflammatory autoimmune diseases, announced today the closing of a $161 million Series B financing. The financing was co-led by Sanofi Ventures, Blue Owl Healthcare Opportunities (formerly Cowen Healthcare Investments), and Norwest Venture Partners, with participation from new investors, B Capital, Leaps by Bayer, Global BioAccess Fund, LOTTE Holdings, Alexandria Venture Investments, and funds managed by abrdn Inc., and existing shareholders, Novo Holdings A/S, Platanus, Bristol Myers Squibb, Digitalis Ventures, and Mission BioCapital.
“We are delighted to partner with additional high-caliber investors. Their scientific and strategic perspectives will support our efforts to diversify our pipeline and bring potentially transformative medicines to patients,” said Pamela Conley, Ph.D., Cofounder, Chief Scientific Officer, and founding Chief Executive Officer of Nuvig Therapeutics.
Nuvig aims to deliver novel methods for reducing autoimmune dysregulation without immunosuppression. Lead candidate NVG-2089, a first-in-class, recombinant, Fc fragment immunomodulator, is engineered to bind type II Fc receptors and engage an endogenous regulatory mechanism that improves autoimmune dysregulation. The proceeds from the Series B financing will support clinical proof-of-concept studies for NVG-2089 and advance Nuvig’s preclinical pipeline. The company is progressing NVG-2089 to Phase 2 clinical development in chronic inflammatory demyelinating polyneuropathy (CIDP) and other undisclosed indications for which there is high unmet need for non-immunosuppressive, efficacious new therapies.
Dosing in the Phase 1 study for NVG-2089 has been successfully completed. In Phase 1 single ascending dose (SAD) and multiple ascending dose (MAD) studies, NVG-2089 was safe and well-tolerated, the pharmacokinetic data were dose proportional, and pharmacodynamic data demonstrated target engagement. The positive results from Phase 1 support the progression of NVG-2089 to Phase 2 studies across multiple indications.
In conjunction with the financing, Paulina Hill, Ph.D., Partner at Sanofi Ventures, Tim Anderson, Managing Director at Blue Owl Capital, and Tiba Aynechi, Ph.D., General Partner at Norwest Venture Partners, joined Nuvig’s Board of Directors.
Dr. Hill said, “Nuvig’s innovative and immunomodulatory approach to inflammatory and autoimmune diseases is well aligned with Sanofi Ventures’ development and investment philosophy. We are impressed by the detailed progress performed by the accomplished team of scientists and executives at Nuvig. They have not only demonstrated powerful anti-inflammatory effects of their lead candidate NVG-2089 without immunosuppression but have also created a reproducible and scalable recombinant method of recapitulating the effects of IVIg without the downsides and supply limitations of IVIg.”
Mr. Anderson added, “We are happy to be a partner to Nuvig on the Series B financing and are very pleased with the recent advancements at Nuvig. In particular, we are excited about Nuvig’s ability to demonstrate that the specific biology of NVG-2089 is engaging the relevant mechanistic pathways in early clinical studies and look forward to further advancing NVG-2089 in the clinic in indications where Nuvig can offer significant advances in safety and efficacy.”
Dr. Aynechi concluded, “We are pleased to support Nuvig’s continued progress towards delivering powerful and safe immune-modulating therapies in autoimmune therapeutic areas where there is significant unmet need. We believe that NVG-2089 has life-changing potential for patients who otherwise have limited treatment options, and Nuvig’s platform programs show promise for applications across a wide range of autoimmune diseases.”
In addition, Nuvig also appointed as independent members of the Nuvig Board of Directors: Ciara Kennedy, Ph.D., founder, President, and CEO of Sorriso Pharmaceuticals, and James Mackay, Ph.D., who most recently was the founder, President, and CEO of Aristea Therapeutics and currently serves as the founder and CEO of Kateran Consulting.
About Chronic Inflammatory Demyelinating Polyneuropathy (CIDP)
Chronic inflammatory demyelinating polyneuropathy (CIDP) is a neurological autoimmune disorder characterized by progressive weakness and impaired sensory function in the legs and arms. Symptoms typically include symmetrical muscle weakness leading to difficulties with walking, climbing stairs, and performing fine motor tasks. Sensory disturbances such as numbness, tingling, or burning sensations. Additionally, diminished or absent reflexes are a common clinical finding. Fatigue is frequent, significantly impacting the patient's quality of life. CIDP symptoms can vary in severity and may progress over several months. Diagnosis is typically confirmed through nerve conduction studies, which reveal demyelination, and cerebrospinal fluid analysis showing elevated protein levels.
About Nuvig Therapeutics
Nuvig Therapeutics is a clinical-stage biotechnology company that is advancing an innovative and transformational pipeline of novel immune therapeutics for chronic inflammatory and autoimmune diseases. The Company’s lead investigational drug candidate, NVG-2089, is an engineered Fc fragment designed to precisely target type II Fc receptors. When NVG-2089 binds to its target, it upregulates the expression of FcgRIIb and causes the expansion of T regulatory cells and the downregulation of numerous inflammatory pathways. Nuvig is based in Menlo Park, California. For more information, please visit www.nuvigtherapeutics.com.
Contacts
Corporate:
info@nuvigtx.com
Media:
Jessica Yingling, Ph.D.,
Little Dog Communications Inc.,
jessica@litldog.com
+1.858.344.8091
Multi-year effort leverages Muna's all-in-human MiND-MAP spatial multi-omics approach to identify and validate new drug targets and treatment pathways for Alzheimer’s disease
GSK secures option to multiple high-value, validated Alzheimer’s-relevant targets for drug discovery, development, and commercialization
Copenhagen, Denmark, December 5, 2024 –Muna Therapeutics (Muna), a biotechnology company focused on developing innovative therapeutics for neurodegenerative diseases, today announced a research alliance with GSK to identify and validate novel drug targets for the treatment of Alzheimer’s disease. The companies will explore insights from Muna’s MiND-MAP platform, which applies spatial transcriptomics to brain samples from Alzheimer’s disease patients, cognitively resilient individuals, healthy controls, and centenarians with and without cognitive impairment. This unique dataset of exceptional breadth and resolution will fuel the discovery and development of innovative medicines for Alzheimer's disease.
Together, Muna and GSK will assess postmortem human brain samples with spatial transcriptomics and other approaches to identify and validate potential new drug targets. The collaboration leverages Muna’s deep expertise in mapping the brain’s response to pathological protein aggregates and its all-in-human platform to identify cellular mechanisms, gene networks, and molecular interactions that underlie brain resilience. Candidate drug targets will be validated using Muna’s suite of humanized cell and animal models, supported by insights from patient tissue and biofluid samples.
“Our agreement marks a pivotal moment in Muna’s evolution and in the broader Alzheimer's research landscape,” said Rita Balice-Gordon, Ph.D., Muna’s Chief Executive Officer. "By combining GSK's commitment to breakthrough science with our MiND-MAP platform's ability to deliver novel insights into brain resilience, we aim to transform the landscape of drug discovery for neurodegenerative diseases and bring new hope to millions of patients worldwide."
Under the terms of the agreement, Muna will receive an upfront payment from GSK of €33.5 million. In addition, Muna will be eligible to receive up to €140 million per target in milestone payments, as well as tiered royalties on net sales of products. Muna will expand and enhance its existing MiND-MAP dataset and will lead the identification and validation of new Alzheimer’s disease targets. GSK will lead drug development and be responsible for preclinical activities, clinical development, manufacturing, and commercialization resulting from work on targets discovered and validated in the collaboration.
“By applying spatial multi-omics to unique patient phenotypes, Muna's MiND-MAP platform is able to determine the genetic and cellular basis of progression and resilience in neurodegenerative diseases,” said Kaivan Khavandi M.D., Ph.D., SVP & Global Head of Respiratory/Immunology R&D at GSK. “The alliance exemplifies our discovery ethos, to utilize advanced data and platform tech to identify high-confidence, human-data-derived, causal targets, which we can support with GSK’s scale and expertise in clinical development and commercialization, to bring desperately needed new therapeutic solutions in Alzheimer’s disease.”
About Muna Therapeutics
Muna Therapeutics discovers and develops therapies that slow or stop devastating neurodegenerative diseases including Alzheimer’s and Parkinson’s disease. These disorders impact memory, movement, language, behavior and personality, resulting in disability and death of millions of patients around the globe. Muna focuses its groundbreaking science on identifying new medicines to preserve cognition and other brain functions, enhance resilience to disease pathology, and slow or stop the progression of neurodegenerative diseases. Its name reflects this focus: Muna means ‘to remember’ in Old Norse. For more information, visit www.munatherapeutics.com. Follow Muna on Linkedin.
###
Media Contact:
Lia Dangelico
Deerfield Group
QRL-101 is the only Kv7 ion channel opener being actively studied for the treatment of hyperexcitability-induced disease progression in ALS, which occurs in nearly 50 percent of patients
Kv7 is a clinically validated target to regulate hyperexcitable state in epilepsy
QurAlis’ Phase 1 study in healthy volunteers of QRL-101 to evaluate biomarkers of both ALS and epilepsy also underway
CAMBRIDGE, Mass., December 4, 2024 – QurAlis Corporation (“QurAlis”), a clinical-stage biotechnology company driving scientific breakthroughs into powerful precision medicines that have the potential to alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative and neurological diseases, today announced that the first patient with ALS has been dosed in a Phase 1 clinical trial evaluating QRL-101 in people living with ALS (QRL-101-04). QRL-101 is a first-in-class selective Kv7.2/7.3 ion channel opener for the treatment of hyperexcitability-induced disease progression in ALS, which is present in approximately 50 percent of ALS patients.
Kv7 hyperexcitability occurs in both sporadic and genetic forms of ALS, with the majority caused by the mis-splicing of the KCNQ2 gene pre-mRNA. Kv7 is also a clinically validated target to regulate the hyperexcitable state in epilepsy. QurAlis recently announced the company has expanded the development program for QRL-101 to include epilepsy.
“QRL-101 is the only Kv7 ion channel opener being actively studied for the treatment of hyperexcitabilityinduced disease progression in ALS. The dosing of the first patient with ALS in the clinical development program of QRL-101 is a significant milestone,” said Kasper Roet, Ph.D., CEO and co-founder of QurAlis. “Kv7 is implicated in ALS as well as epilepsy. We believe that the data from this study, along with the data from our Phase 1 study evaluating biomarkers of ALS and epilepsy in healthy volunteers, will be valuable as we advance the clinical program for QRL-101 in ALS so that we can bring a much-needed therapeutic option to patients rapidly.”
“Preclinical models of QRL-101 show its strong potential to control motor neuron hyperexcitabilityinduced neurodegeneration with an attractive side effect profile,” said Leonard H. van den Berg, M.D., Ph.D., professor of neurology and chair, TRICALS. “ALS is a devastating, fatal neurodegenerative disease and there are currently no therapies that can significantly extend patients’ lives. We look forward to results from this Phase 1 study in ALS patients.”
QRL-101-04 (NCT06714396) is a Phase 1 proof-of-mechanism (PoM) single-dose, placebo-controlled clinical trial designed to evaluate the safety and tolerability of QRL-101 in people living with ALS. The study is expected to enroll approximately 12 participants with ALS and will evaluate the impact of QRL-101 on excitability biomarkers including on the strength-duration time constant (SDTC), a known predictor of survival in ALS patients. QurAlis is also conducting a Phase 1 PoM biomarker clinical trial (QRL-101-05; NCT06681441) to evaluate biomarkers of ALS and epilepsy of QRL-101 in healthy volunteers. These PoM studies together will inform QurAlis’ future development program, including dose levels for proof-of-concept studies.
QurAlis anticipates reporting topline data from the QRL-101-04 Phase 1 clinical trial in the first half of 2025.
More information about the QRL-101 Phase 1 clinical trials can be found at www.clinicaltrials.gov.
About Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a progressive neurodegenerative disease impacting nerve cells in the brain and spinal cord, reducing muscle function and control. ALS can be traced to mutations in more than 25 different genes and is often caused by a combination of multiple sub-forms of the condition. Cases usually cannot be predicted, although a small percentage are inherited. ALS has a devastating impact on patients and families. ALS patients’ average life expectancy is three years; after diagnosis patients only have two years to live. There is currently no cure for the disease.
About Kv7
Kv7.2/7.3 is a voltage-gated potassium channel whose role is crucial for the regulation of neuronal excitability and membrane potential. Kv7.2 is mis-spliced in sporadic amyotrophic lateral sclerosis (ALS) leading to loss of function and abnormal electrical activity in the spinal cord and brain. The activation of this channel shows the potential to decrease spinal and cortical/motor neuron excitability and to positively affect CMAP (compound muscle action potential), a disease progression biomarker for ALS. This suggests that this may be an effective therapeutic approach for ALS patients suffering from hyperexcitability-induced motor neuron degeneration.
About QurAlis Corporation
At QurAlis, we are neuro pioneers on a quest to cure. We work with a relentless pursuit of knowledge, a precise attention to craft, and an optimistic mindset to discover and develop effective precision medicines that have the potential to alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative and neurological diseases. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a robust precision medicine pipeline with therapeutic candidates aimed at modifying severe disease pathology in defined patient populations based on both disease-causing genetic mutation(s) and clinical biomarkers. For more information, please visit www.quralis.com or follow us on X @QurAlisCo or LinkedIn.
Contact:
Kathy Vincent
kathy.vincent@quralis.com
310-403-8951
- SUDO-550 is a potential best-in-class, orally administered, brain-penetrant TYK2 inhibitorbeing developed for the treatment of neuroinflammatory diseases.
- Phase 1 clinical trial supported by preclinical data demonstrating high potency, selectivity,and efficacy in preclinical models, along with the ability to cross the blood-brain barrier.
CARMEL, Ind.--(BUSINESS WIRE)-- Sudo Biosciences (“Sudo”), a biopharmaceutical company committed to designing and developing best-in-class precision TYK2 (tyrosine kinase 2) inhibitors, announced today that the first participants have been dosed in a Phase 1 clinical trial evaluating SUDO-550, a novel brain-penetrant allosteric TYK2 inhibitor for the treatment of neuroinflammatory diseases.
The Phase 1 clinical trial is designed to evaluate the safety, tolerability, and pharmacokinetics of single and multiple- ascending doses of SUDO-550 in healthy volunteers, including confirmation that the compound effectively crosses the blood-brain barrier.
“Entering the clinic is a critical step in the development of SUDO-550 and establishing it as a best-in-class brain-penetrant TYK2 inhibitor. This is a therapy that could significantly advance the treatment of diseases such as multiple sclerosis, ALS and Alzheimer’s,” said Ian Mills, Chief Medical Officer, Sudo Biosciences. “This is the second allosteric TYK2 inhibitor we have advanced into the clinic this year, after SUDO-286 which is progressing in two Phase 1 trials as a topical treatment for psoriasis.”
The company is developing multiple highly potent and selective small molecule TYK2 pseudokinase inhibitors designed to provide targeted treatments across a broad range of autoimmune and neurologic conditions.
About SUDO-550
SUDO-550 is an orally administered, allosteric TYK2 inhibitor that demonstrates high selectivity and potency for TYK2, minimizing off-target effects. Non-clinical studies have demonstrated excellent blood-brain barrier penetration with the compound, enabling therapeutic potential for CNS diseases characterized by compartmentalized neuroinflammation. These results support its potential as a best-in-class treatment for multiple neuroinflammatory diseases.
About SUDO-286
SUDO-286 is a highly potent, selective and potential first and best-in-class topical TYK2 inhibitor for psoriasis and other immune-mediated dermatologic diseases. The program entered the clinic earlier this year and is currently being evaluated in two Phase 1 studies in healthy volunteers and patients.
About Sudo Biosciences
Sudo Biosciences is a biopharmaceutical company committed to designing and developing novel medicines to transform patients’ lives. The company’s programs target the tyrosine kinase 2 (TYK2) pseudokinase domain. TYK2 is a key mediator in cytokine signaling pathways that have been linked to a broad range of immune-mediated inflammatory conditions. The company’s pipeline of next generation TYK2 inhibitors includes a potential first- and best-in-class brain- penetrant candidate for the treatment of multiple sclerosis and neurodegenerative diseases with underlying neuroinflammation and a potential first- and best-in-class topical candidate for immune-mediated dermatologic diseases. Sudo Biosciences is based in Carmel, IN, with operations across the US and UK. For more information, visit www.sudobio.com.
Contacts Media
Kimberly Ha
KKH Advisors
917-291-5744
kimberly.ha@kkhadvisors.com
Source: Sudo Biosciences
- PK data analysis in dose-escalation phase indicated exposures of QRL-201 met or exceeded the targeted
- therapeutic range prompting advancement to the DRF phase
- ANQUR protocol amended to include additional biomarkers and cohort of participants with C9orf72-related ALS; Company to present update at 35th International Symposium on ALS/MND
CAMBRIDGE, Mass., Nov. 19, 2024 /PRNewswire/ -- QurAlis Corporation, a clinical-stage biotechnology company driving scientific breakthroughs into powerful precision medicines that have the potential to alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative and neurological diseases, today announced that the Phase 1 ANQUR clinical trial evaluating QRL-201 for the treatment of ALS has successfully completed the dose-escalation phase, based on pharmacokinetic (PK) data analysis from Cohorts 1 and 2, indicating cerebrospinal fluid (CSF) exposure levels of QRL-201 met or exceeded the targeted therapeutic range. QRL-201 is a first-in-class precision therapeutic product candidate that has the potential to restore STATHMIN-2 (STMN2) expression in ALS patients with the aim to modify disease progression and improve outcomes.
The ANQUR clinical trial (QRL-201-01; NCT05633459) has advanced to the dose range-finding (DRF) phase which will evaluate two doses of QRL-201 and include additional biomarkers. The study design will now also include a cohort of participants with C9orf72-related ALS in addition to participants with sporadic ALS. Based on previous expression analysis, patients with C9orf72-related ALS show consistent mis-splicing of STMN2. The ANQUR clinical trial is currently active in Canada, the United Kingdom (UK), and the European Union (EU). Cohorts 1 and 2 have successfully completed dosing and an amendment to the DRF study design has been approved in Canada and the UK, with anticipated approval from the EU in the near future. The first participant in the DRF phase has been dosed in Canada.
"ALS is a devastating, fatal neurodegenerative disease with a large unmet medical need. Our mission is to make a meaningful difference in patients' lives, and we believe QRL-201 could potentially modify disease progression and improve outcomes in ALS patients who have a loss of STMN2 due to TDP-43 pathology," said Kasper Roet, Ph.D., CEO and co-founder of QurAlis. "The PK data analysis from the first two completed cohorts of ANQUR indicated CSF exposures of QRL-201 met or exceeded the targeted therapeutic range. These findings further support our confidence in the potential therapeutic effect of QRL-201 in the treatment of sporadic ALS."
STMN2 is a well-validated protein important for neural repair, axonal stability, and muscle innervation and is the most significantly regulated gene by TDP-43 exclusively in humans. STMN2 is the most consistently decreased gene across multiple ALS RNA expression studies. Loss of nuclear TDP-43 leads to mis-splicing of the pre-mRNA of STMN2, resulting in loss of full-length transcript and protein. QRL-201 rescues STMN2 loss of function in QurAlis' ALS patient-derived motor neuron disease models in the presence of TDP-43 pathology. TDP-43 pathology is associated with nearly all ALS patients, approximately 50 percent of patients with FTD, the second most common form of dementia, and with about a third of Alzheimer's Disease patients. There are currently no cures for ALS or FTD, and there are limited therapeutic options available for ALS and FTD patients who are in desperate need of effective therapies.
"We are pleased that the first participant in Canada has been dosed in the dose range-finding stage of the ANQUR clinical trial," said Doug Williamson, M.D., QurAlis' chief medical officer. "This represents a significant milestone in the QRL-201 program to evaluate a potential transformative breakthrough precision medicine for patients with sporadic ALS."
QurAlis will present an update on the ANQUR clinical trial in a poster presentation at the 35th International Symposium on ALS/MND being held December 6-8, 2024, in Montreal, Canada and virtually. Details of the presentation are as follows:
Title: QRL-201-01 (ANQUR): A multicenter, randomized, double-blind, placebo-controlled multiple ascending dose study to evaluate the safety and tolerability of QRL-201 in amyotrophic lateral sclerosis
Date/Time: Saturday, December 7, 2024 5:00-7:00PM ET
Theme: Clinical Trials and Trial Design
Abstract Number: CLT-11
Session: Poster Session B
Presenter: Emma Bowden, Ph.D., senior vice president, head of clinical development, QurAlis
About the ANQUR Clinical Trial
ANQUR (QRL-201-01; NCT05633459) is the first-ever clinical trial to evaluate a potential therapy to rescue STATHMIN-2 (STMN2) expression in people with amyotrophic lateral sclerosis (ALS). ANQUR is a global, multi-center, randomized, double-blind, placebo-controlled multiple-ascending dose Phase 1 clinical trial designed to evaluate the safety and tolerability of QRL-201 versus placebo in participants with ALS. The primary objective and endpoint of the study is to determine the safety and tolerability of multiple doses of QRL-201. The secondary objective and endpoint is the plasma pharmacokinetic (PK) profile of QRL-201 after multiple doses. ANQUR also intends to evaluate multiple exploratory endpoints including biomarkers of neuronal loss and STMN2 biology (neurofilament levels, STMN2 levels, Chitinase-3-like protein 1, and miRNA profiles), clinical outcome measures (ALSFRS-R, ALSAQ-5, ROADS, King's Staging, SVC, muscle strength, electrophysiology markers of denervation [maximum compound muscle action potential/CMAP and repetitive nerve stimulation/RNS]), and cerebrospinal fluid (CSF) PK profile.
The ANQUR clinical trial is expected to include 64 study participants with ALS across sites in Canada, the European Union (EU), and United Kingdom (UK). Sites participating in the ANQUR clinical trial: Canada – University of Alberta (Edmonton, Alberta) University of Calgary (Calgary, Alberta), Montréal Neurological Institute-Hospital (Montreal, Quebec), and CHUM-Hopital Notre-Dame (Montréal, Quebec); EU – Universitaire Ziekenhuizen Leuven (Leuven, Belgium), Charité Research Organisation (Berlin, Germany), University Hospital Schleswig-Holstein Campus Lübeck (Lübeck, Germany), Universitätsklinikum Ulm (Ulm, Germany), Universitair Medisch Centrum Utrecht (Utrecht, The Netherlands); and UK – St. James Hospital (Dublin, Ireland), Kings College Hospital NHS Foundation Trust (London, England), and The University of Sheffield, Royal Hallamshire Hospital (Sheffield, England).
The ANQUR clinical trial successfully completed the dose-escalation phase, based on PK data analysis from Cohorts 1 and 2, indicating CSF exposure levels of QRL-201 met or exceeded the targeted therapeutic range. The dose range-finding phase will evaluate two doses of QRL-201 and will enroll an additional 48 participants: 32 participants with sporadic ALS and 16 participants with C9orf72-related ALS.
Visit www.clinicaltrials.gov for more information about the ANQUR study.
About QurAlis Corporation
At QurAlis, we are neuro pioneers on a quest to cure. We work with a relentless pursuit of knowledge, a precise attention to craft, and an optimistic mindset to discover and develop effective precision medicines that have the potential to alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative and neurological diseases. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a robust precision medicine pipeline with therapeutic candidates aimed at modifying severe disease pathology in defined patient populations based on both disease-causing genetic mutation(s) and clinical biomarkers. For more information, please visit www.quralis.com or follow us on X @QurAlisCo or LinkedIn.
Media contact:
Kathy Vincent
kathy@kathyvincent.com
310-403-8951
- Aliada's lead compound, ALIA-1758, an anti-pyroglutamate amyloid beta (3pE-Aβ) antibody, is a potential best-in-class therapy for Alzheimer's disease
- Acquisition also allows AbbVie to utilize Aliada's novel blood-brain barrier (BBB)-crossing technology to enhance discovery and development efforts across neuroscience
NORTH CHICAGO, Ill. and BOSTON, Oct. 28, 2024 /PRNewswire/ -- AbbVie (NYSE: ABBV) and Aliada Therapeutics today announced a definitive agreement under which AbbVie will acquire Aliada, a biotechnology company advancing therapies using a novel blood-brain barrier (BBB)-crossing technology to address challenging central nervous system (CNS) diseases. Aliada's lead investigational asset utilizing this delivery technology, ALIA-1758, is an anti-pyroglutamate amyloid beta (3pE-Aβ) antibody in development for the treatment of Alzheimer's disease.
"Neuroscience is one of our key growth areas and we are committed to driving innovation in this field to address critical unmet needs for patients living with seriously debilitating neurological diseases such as Alzheimer's disease," said Roopal Thakkar, M.D., executive vice president, research and development and chief scientific officer, AbbVie. "This acquisition immediately positions us to advance ALIA-1758, a potentially best-in-class disease-modifying therapy for Alzheimer's disease. In addition, Aliada's novel BBB-crossing technology strengthens our R&D capabilities to accelerate the development of next-generation therapies for neurological disorders and other diseases where enhanced delivery of therapeutics into the CNS is beneficial."
"We are pleased to announce the acquisition of Aliada by AbbVie and are excited about AbbVie's commitment to bringing ALIA-1758 to patients with Alzheimer's disease. Our proprietary MODEL™ platform has enabled the development of ALIA-1758, a promising step forward in brain delivery of an anti-amyloid antibody therapy," said Michael Ryan, M.D., chief medical officer at Aliada Therapeutics. "Many promising CNS-targeted therapies fail to reach late-stage trials due to their inability to cross the blood-brain barrier. Our MODEL™ platform addresses this challenge directly, efficiently delivering targeted drugs and potentially transforming how we treat neurological diseases."
Aliada is advancing therapeutic candidates using its Modular Delivery (MODEL™) platform, engineered for high-precision CNS drug delivery. The novel BBB-crossing technology targets transferrin and CD98 receptors (TfR and CD98) which are highly expressed in brain endothelial cells. By engineering highly optimized TfR or CD98 binders, this platform is designed to deliver different types of biological cargoes into the brain, including therapeutic antibodies and genetic medicines such as siRNA.
ALIA-1758 utilizes TfR to transport a 3pE-Aβ antibody across the BBB to enable degradation and elimination of amyloid beta plaques, a pathological hallmark of Alzheimer's disease. This investigational candidate is currently in a Phase 1 clinical trial to assess its safety and tolerability in healthy participants (NCT06406348).
Under the terms of the agreement, AbbVie will acquire all outstanding Aliada equity for $1.4 billion in cash, subject to certain customary adjustments. This transaction is expected to close in 4Q2024, subject to regulatory approvals and other customary closing conditions.
Advisors
AbbVie's legal advisor was Covington & Burling LLP. Aliada's exclusive financial advisor was Centerview Partners LLC and Fenwick & West LLP served as legal advisor.
About AbbVie
AbbVie's mission is to discover and deliver innovative medicines and solutions that solve serious health issues today and address the medical challenges of tomorrow. We strive to have a remarkable impact on people's lives across several key therapeutic areas – immunology, oncology, neuroscience, and eye care – and products and services in our Allergan Aesthetics portfolio. For more information about AbbVie, please visit us at www.abbvie.com. Follow @abbvie on LinkedIn, Facebook, Instagram, X (formerly Twitter), and YouTube.
About Aliada Therapeutics
Aliada Therapeutics is a biotechnology company focused on addressing delivery challenges in CNS drug development. Aliada is developing next-generation CNS therapeutics with its novel BBB-crossing Modular Delivery (MODEL™) platform technology, which has been shown to efficiently transport diverse therapeutic cargoes into the brain, enhancing effectiveness and addressing the critical need for efficient and versatile large molecule and oligonucleotide delivery. Johnson & Johnson (through its venture capital arm, Johnson & Johnson Innovation – JJDC, Inc.), RA Capital Management, and Raven (RA Capital Management's incubator) co-founded Aliada and co-led the series seed financing in 2021 to advance the MODEL™ platform created by Johnson & Johnson scientists that was licensed to Aliada at its inception. Further investment was made by OrbiMed and Sanofi Ventures.
For more information visit www.aliadatx.com and follow us on LinkedIn and X.
Forward-Looking Statements
Some statements in this news release are, or may be considered, forward-looking statements for purposes of the Private Securities Litigation Reform Act of 1995. The words "believe," "expect," "anticipate," "project" and similar expressions and uses of future or conditional verbs, generally identify forward-looking statements. AbbVie cautions that these forward-looking statements are subject to risks and uncertainties that may cause actual results to differ materially from those expressed or implied in the forward-looking statements. Such risks and uncertainties include, but are not limited to, challenges to intellectual property, competition from other products, difficulties inherent in the research and development process, adverse litigation or government action, and changes to laws and regulations applicable to our industry. Additional information about the economic, competitive, governmental, technological and other factors that may affect AbbVie's operations is set forth in Item 1A, "Risk Factors," of AbbVie's 2023 Annual Report on Form 10-K, which has been filed with the Securities and Exchange Commission, as updated by its subsequent Quarterly Reports on Form 10-Q. AbbVie undertakes no obligation, and specifically declines, to release publicly any revisions to forward-looking statements as a result of subsequent events or developments, except as required by law.
- Dr. Gaspar brings invaluable expertise to gene-editing pioneering company Eligo as it prepares for clinical trials with world’s first topical CRISPR treatment
- Eligo also strengthens its investor network with new strategic investors, including VIVES Partners and CRISPR pioneer Rodolphe Barrangou
Paris, France, October 1, 2024 – Eligo Bioscience, a biotech company expanding the scope of gene-editing targets by focusing on the delivery of genetic medicines to the microbiome, today announces the appointment of Dr. Bobby Gaspar, CEO of Orchard Therapeutics, as chair of its board of directors. This announcement follows the publication in the prestigious Nature journal, highlighting its novel approach to using gene editing to target the microbiome.
“I am thrilled to welcome Bobby as chair of Eligo. His exceptional ability to translate breakthrough innovations from concept to commercialization at Orchard Therapeutics is impressive. As a co-founder and CEO of a pioneering genetic medicines company, his expertise will be invaluable as Eligo advances toward clinical trials and expands its therapeutic pipeline,” said Xavier Duportet, CEO and co-founder of Eligo.
With three decades of experience spearheading the development of cutting-edge gene therapies, Dr. Bobby Gaspar is a recognized pioneer in genetic medicine. As the co-founder and CEO of Orchard Therapeutics, his leadership guided the company from early development stages through regulatory approvals and commercialization in Europe and the US, as well as a notable $477M acquisition in 2023. An honorary professor of pediatrics and immunology at University College London, Dr. Gaspar was named on the inaugural TIME100 Health List, recognizing him as one of the world’s most influential individuals in health. His extensive expertise in both private and public fundraising, along with a proven track record of taking therapies from lab to market, will greatly benefit Eligo as it enters this critical next phase.
“In my early career as a pediatrician caring for children with devastating genetic diseases, I developed a passion and commitment for transforming the way medicine is practiced through the delivery of paradigm-shifting therapies,” said Dr. Gaspar. “At Eligo, there is an unprecedented opportunity to utilize gene editing technology to make specific and targeted genetic changes to patients’ microbiome and address a wide range of severe diseases. What the company has achieved since its inception is impressive, and I look forward to partnering with Xavier and the broader team to help translate this powerful platform into new scientific breakthroughs and innovative treatments.”
Eligo Expands Investor Syndicate with Key Strategic Additions
Eligo is pleased to welcome new investors, VIVES Partners, which is supported by the European Union under the InvestEU Fund, and Prof. Rodolphe Barrangou, a CRISPR pioneer and co-founder of Intellia Therapeutics. The continued interest from these strategic investors, following the company’s successful $30M fundraising round last year, underscores the strong potential of Eligo’s first-in-class gene-editing platform.
“This is a very exciting time for translational pursuits of CRISPR-based effectors into the clinic and I am delighted to support Eligo’s journey as it gets to the clinical stage of in vivo editing of the microbiome and disrupts the field with a radically game-changing and innovative approach,” said Rodolphe Barrangou.
Leveraging its proprietary platform, Eligo is developing first-in-class modalities that deliver CRISPR and therapeutic genetic circuits directly to the microbiome, enabling precise editing of both its genetic composition and function. Eligo is progressing toward a key milestone: advancing its lead program into clinical trials for moderate to severe acne with the world’s first topical CRISPR treatment.
Moderate to severe acne affects approximately 120 million patients globally, with a quality-of-life impact comparable to conditions like asthma, diabetes and epilepsy. Despite the significant unmet need, innovation in acne treatment has been almost stagnant for the past three decades. Eligo’s novel mechanism of action, targeting microbiome dysbiosis through the CRISPR-based precise elimination of pro-inflammatory C. acnes strains within hair follicles, offers a groundbreaking and targeted solution. This represents a significant opportunity for safe, effective treatment in a $10 billion market ripe for disruption.
“In addressing the pressing medical need for effective solutions to severe acne, we recognize that current treatments fall short, often accompanied by significant adverse effects and limited efficacy. This large unmet medical need profoundly impacts a young population, struggling with not only physical skin defects but also psychological challenges. That’s why we are committed to investing in Eligo and collaborating with a strong consortium of investors, as we believe its pioneering approach holds the potential to truly tackle this critical issue,” said Philippe Durieux, CEO of VIVES Partners.
About Eligo Bioscience
Eligo Bioscience is the world leader in microbiome in vivo gene editing and is advancing a highly differentiated pipeline of precision medicines to address unmet medical needs in immunoinflammation, oncology and infectious diseases driven by the expression of deleterious bacterial genes.
Eligo was founded by Luciano Marraffini (Professor at The Rockefeller University and co-founder of Intellia Therapeutics), Timothy Lu (Professor at MIT, and CEO at Senti Biosciences), Dr. David Bikard (Professor at Institut Pasteur) and Xavier Duportet (MIT TR35, Young Global Leader, and Termeer Fellow).
Eligo was named a Technology Pioneer by the World Economic Forum and received venture capital funding from Sanofi Ventures, Khosla Ventures, Bpifrance, Seventure Partners and VIVES Partners.
Media and analyst contact
Andrew Lloyd & Associates
Juliette Schmitt & Saffiyah Khalique
juliette@ala.associates - saffiyah@ala.associates
UK/US: +44 1273 952 481
Oxford, United Kingdom – 30 September 2024 – OMass Therapeutics (‘OMass’ or ‘the Company’), a biotechnology company identifying medicines against highly validated target ecosystems such as membrane proteins or intracellular complexes, today announces the candidate nomination of its lead program targeting the melanocortin-2 (MC2) receptor and key appointments to support its progress towards becoming a clinical-stage company.
Over the last year, OMass has made significant progress in advancing its pipeline and has selected its first clinical candidate targeting the MC2 receptor, a GPCR for the adrenocorticotropic hormone (ACTH). The focus of the program has been to increase the receptor residency time to make OMass’ antagonists resistant to competition by the endogenous ligand. This can allow all patients suffering from conditions related to ACTH excess, including congenital adrenal hyperplasia and Cushing’s syndrome, to be treated. The long residence time is expected to avoid loss of efficacy in the face of rising ACTH levels due to reductions in glucocorticoid supplementation for CAH or progression of Cushing’s Syndrome.
To support the advancement of OMass’ pipeline, OMass has expanded its development team. Based in the USA, Dr Steve Griffen joins as Vice President of Clinical Development and Angela Hecyk joins as Director of Clinical Operations. Based in the UK, Stuart Hadley has been appointed as Senior Director of Chemistry Manufacture and Controls (CMC). The new appointments add critical expertise in clinical development and manufacturing:
- Steve is an endocrinologist with more than 25 years’ experience in basic and clinical research and drug development. He has previously served as Vice President, Clinical Development at MBX Biosciences following his role as the Medical Lead for Integrated Care for Sanofi developing devices and software to assist people managing their diabetes. Prior to that, he served as Senior Vice President, Research for the non-profit organization JDRF and Type 1 Diabetes Full Development Team leader for dapagliflozin at Bristol-Myers Squibb, after having served as Exploratory Development Team leader for metabolic assets taking multiple assets into first-in-human studies. Steve holds an AB in Physiology and an MA in Endocrinology from the University of California, Berkeley and completed his medical doctorate at the Medical College of Wisconsin.
- Angela is a global clinical operations professional with over 16 years of experience in clinical research across all stages of clinical development to post-approval, including significant contributions to programs in rare pediatric diseases and four FDA-approved therapies. She joins OMass after holding positions at several growing small to mid-sized biotech and pharmaceutical companies, including MBX Biosciences, Taysha Gene Therapies, Ayala Pharma, Phathom Pharmaceuticals, and Astellas Pharma. Prior to this, she oversaw clinical trials in islet cell transplantation at Northwestern University and was a study monitor for ICON. Angela holds a BS in Natural Sciences, Biology from the University of Wisconsin-Madison.
- Stuart has over 25 years' experience in drug development and operations working across all disciplines of CMC and supply chain management. He joins OMass from F2G Ltd, where he was Senior Director CMC, leading the CMC program for Olorofim to launch readiness. Prior to that he held multiple CMC and supply chain roles over a 20-year period with AstraZeneca, starting his career there as a graduate analytical chemist, having completed his BSc (Hons) in Chemistry with Pharmaceutical and Forensic Science at the University of Bradford.
Ros Deegan, Chief Executive Officer at OMass Therapeutics, commented: “Nomination of our first clinical candidate represents a key milestone for the company. I am delighted to welcome Steve, Angela and Stuart to the OMass team. They bring extensive experience, and I have no doubt that they will prove to be invaluable additions as we progress our MC2 program to the clinic.”
Steve Griffen, Vice President of Clinical Development at OMass Therapeutics, added: “I’m very excited by the opportunity to join OMass at such a pivotal time for the company. I look forward to working closely with Ros, Stuart, Angela and the rest of the OMass team in advancing our pipeline of small molecule drug candidates.”
-ENDS-
For further information, please contact:
|
OMass Therapeutics |
ICR Consilium |
|
Rosamond Deegan, Chief Executive Officer Phone: +44 (0) 1235 527589 Email: ros.deegan@omass.com
|
Sue Charles / Suki Virji / Kumail Waljee Phone: +44 (0)20 3709 5700 Email: omass@icrhealthcare.com
|
About OMass Therapeutics
OMass Therapeutics is a biotechnology company discovering medicines against highly-validated target ecosystems, such as membrane proteins or intracellular complexes.
OdyssION™, OMass’ unique drug discovery platform, comprises next-generation native mass spectrometry with novel biochemistry techniques and custom chemistry to interrogate not just a drug target, but also the interaction of the target with its native ecosystem, separate from the confounding complexity of the cell. This unique approach results in cell-system fidelity with cell-free precision.
OMass is advancing a pipeline of small molecule therapeutics in rare diseases and immunological conditions. Its lead programme is a best-in-class MC2 (melanocortin-2) receptor antagonist for the treatment of Congenital Adrenal Hyperplasia (CAH) and Cushing’s Syndrome. The focus of the program has been to increase receptor residency time to make OMass’ antagonists resistant to competition by the endogenous ligand, thereby avoiding loss of efficacy in the face of rising adrenocorticotropic hormone (ACTH) levels due to reductions in glucocorticoid supplementation for CAH or progression of Cushing’s Syndrome.
Headquartered in Oxford, UK, OMass has raised over $160M (£129M) from a top-tier international investor syndicate including Syncona, Oxford Science Enterprises, GV, Northpond Ventures, Sanofi Ventures and British Patient Capital.
To learn more, please visit www.omass.com. Follow us on LinkedIn and X.
- QRL-101 is the only Kv7.2/7.3 ion channel opener being actively studied for the treatment of hyperexcitability-induced disease progression in ALS
- Kv7.2/7.3 is a clinically validated target to regulate the hyperexcitable state in epilepsy
- Company initiates exploratory Phase 1 proof-of-mechanism study in healthy volunteers to characterize the potential anti-seizure effects of QRL-101
CAMBRIDGE, Mass., September 19, 2024 – QurAlis Corporation (“QurAlis”), a clinical-stage biotechnology company driving scientific breakthroughs into powerful precision medicines that have the potential to alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative and neurological diseases, today announced that the company is expanding its Kv7 development program to include epilepsy. The Company’s lead investigational candidate, QRL-101 is the only Kv7.2/7.3 ion channel opener being actively studied for the treatment of hyperexcitability-induced disease progression in ALS, which is present in approximately 50 percent of ALS patients. Kv7 is a clinically validated target to regulate the hyperexcitable state in epilepsy.
QurAlis also announced the initiation of an exploratory Phase 1 proof-of-mechanism electroencephalogram biomarker study in healthy volunteers of QRL-101 to characterize the potential anti-seizure effects of QRL-101.
“Epilepsy, one of the most common and most disabling neurological seizure disorders, is characterized by spontaneous recurrent seizures, which disrupt normal brain functions, lead to neuronal loss, and result in cognitive and emotional deficits. In about one-third of people living with epilepsy, the seizures are resistant to current treatments; so more effective treatments are urgently needed,” said Kasper Roet, Ph.D., chief executive officer and co-founder of QurAlis. “QRL-101, is a highly selective Kv7.2/7.3 ion channel opener, which in preclinical models shows a strong potential to control motor neuron hyperexcitability-induced neurodegeneration with an attractive side effect profile. Since Kv7 is a clinically validated target in controlling hyperexcitability in epilepsy, we are excited to expand our scope of QRL-101 into a new therapeutic area and explore the potential of QRL-101 in epilepsy so that QurAlis can continue the goal of making a real difference in patients’ lives.”
About Kv7
Kv7.2/7.3 is a voltage-gated potassium channel whose role is crucial for the regulation of neuronal excitability and membrane potential. The activation of this channel shows the potential to decrease spinal and cortical motor neuron excitability and to positively affect several electrophysiological biomarkers. This suggests that this may be an effective therapeutic approach in several neurodegenerative and neurological diseases including ALS and epilepsy.
About Epilepsy
Epilepsy is one of the most common chronic neurological diseases and, according to the Centers for Disease Control, affects more than 65 million people around the world of which 3.4 million are in the U.S. Epilepsy is characterized by unpredictable, recurrent seizures, which are brief episodes of involuntary movement that may involve a part of the body (partial) or the entire body (generalized). Seizure episodes
are a result of excessive electrical discharges in a group of brain cells. According to the World Health Organization, recurrent seizures disrupt normal brain functions, lead to neuronal loss, and result in cognitive and emotional deficits. Patients suffer from stigmatization, social isolation, combined with disability, educational underachievement, and poor employment outcomes. The Epilepsy Foundation estimates that one-third of people with epilepsy live with uncontrollable seizures because no available treatments are effective.
About QurAlis Corporation
At QurAlis, we are neuro pioneers on a quest to cure. We work with a relentless pursuit of knowledge, a precise attention to craft, and an optimistic mindset to discover and develop effective precision medicines that have the potential to alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative and neurological diseases. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a robust precision medicine pipeline with therapeutic candidates aimed at modifying severe disease pathology in defined patient populations based on both disease-causing genetic mutation(s) and clinical biomarkers. For more information, please visit www.quralis.com or follow us on X @QurAlisCo or LinkedIn.
Contact: Kathy Vincent kathy.vincent@quralis.com 310-403-8951

- Proceeds will support Nura Bio’s pipeline of neuroprotective medicines
- Company announces successful completion of Phase 1 study of NB-4746, Nura Bio’sbrain-penetrant SARM1 inhibitor
- Shilpa Sambashivan, Ph.D., appointed CEO and a Director of the company
South San Francisco, Calif., September 17, 2024--(BUSINESS WIRE)- Nura Bio Inc. (Nura Bio), a clinical- stage, biopharmaceutical company developing neuroprotective, small molecule therapies for the treatment of debilitating neurological diseases, announced today the closing of more than $140 million in Series A financing. This includes the addition of $68 million to the initial Series A round of $73 million which was announced in 2020. The round was led by founding investor The Column Group, with participation from continuing investors Samsara Bio Capital and Euclidean Capital, and new investor Sanofi Ventures.
The company also announced the appointment of Shilpa Sambashivan, Ph.D., as Chief Executive Officer (CEO) and a company Director. As a member of the founding team at Nura Bio and Chief Scientific Officer (CSO), Dr. Sambashivan has been the driving force behind Nura Bio’s bespoke research engine and differentiated R&D pipeline, with the company’s first clinical candidate, NB-4746, recently completing Phase 1 studies in healthy volunteers.
“Under Shilpa’s leadership, Nura Bio has successfully transitioned to a clinical-stage organization, making remarkable progress in identifying ways to translate complex biology into potential therapies,” said Tim Kutzkey, Ph.D., Managing Partner, The Column Group and Nura Bio’s founding chairman. “We are excited to continue to support the company through this next phase of growth and clinical development. Shilpa’s leadership, combined with her deep scientific expertise, will be key to maximizing Nura Bio’s broad therapeutic potential in areas of large unmet need.”
Nura Bio’s financing close comes at a pivotal point with the Phase 1 success of NB-4746, a brain- penetrant SARM1 inhibitor that has been shown to prevent axon degeneration and provide neuroprotection in multiple preclinical models of nerve injury and disease. Nura Bio plans to initiate a Phase 1b/2 trial in a patient population in 2025.
“At Nura Bio, we have been laser-focused on our mission of delivering novel neuroprotective therapies to patients by leveraging our deep scientific understanding of underlying disease mechanisms including axon degeneration and neuroinflammation. The strong support demonstrated by our investors through this financing reflects the tremendous potential of our R&D pipeline,” said Dr. Sambashivan. “I am proud of the results our team has delivered. I look forward to leading the company through this next phase as we prepare to test the SARM1 hypothesis in a patient population in 2025 with our lead candidate NB-4746 while continuing to advance our promising preclinical pipeline.”
Results from the recently completed Phase 1 study of NB-4746 in healthy volunteers show it was well-tolerated in the single ascending and multiple ascending dose arms of the study. In this Phase 1 study, NB-4746 achieved targeted plasma exposure levels that the company believes are required for efficacy with no associated serious treatment-emergent adverse events. Cerebrospinal fluid levels of NB-4746 confirm brain penetration and support this molecule's advancement in diseases that impact both the peripheral and central nervous systems.
About NB-4746
NB-4746 is the lead asset in Nura Bio’s small molecule pipeline. NB-4746 targets SARM1, a neuronally enriched nicotinamide adenine dinucleotide (NAD) hydrolase that has emerged as an important axon- intrinsic metabolic sensor and central driver of axon degeneration. Axon degeneration is an early hallmark of several neurological diseases. Halting axon degeneration early can confer significant structural and functional neuroprotection and has tremendous potential in the treatment of several neurological diseases. Preclinical studies support the potential of NB-4746 to provide broad axonal protection and functional improvement across diseases of the central, peripheral, and ocular nervous systems.
About Nura Bio
Nura Bio, Inc. (Nura Bio) is a clinical-stage biopharmaceutical company developing neuroprotective therapies for the treatment of a broad range of neurological diseases. Nura Bio’s research and early development small molecule pipeline is focused on developing therapies that halt axon degeneration and/or modulate microglial responses to degeneration and injury, with the goal of conferring neuroprotection, across diseases of the central, peripheral, and ocular nervous systems.
- MinervaX scaling up supply of novel GBS vaccine, ahead of phase III studies
- Wacker Biotech to manufacture active vaccine protein ingredients and prepare for commercial supply after regulatory approval
- Vaccine to address the unmet medical burden of GBS, both by maternal vaccination to prevent adverse pregnancy outcomes and life-threatening infections in infants and by vaccination of older/at risk adults
Copenhagen/Munich, September 17, 2024 – MinervaX ApS, a privately held Danish biotechnology company developing a novel, prophylactic vaccine against Group B Streptococcus (GBS) and Wacker Biotech, a contract development and manufacturing organization (CDMO), wholly owned subsidiary of Wacker Chemie AG, today announce a collaboration to manufacture MinervaX’s active protein ingredients of its GBS vaccine.
GBS is responsible for nearly 50 percent of all life-threatening infections in newborns. At any given time, some 15-25 percent of the population, including pregnant people are spontaneously colonized with GBS, and during pregnancy they run the risk of transmitting the bacteria to their child in the womb, during birth and/or during the first months of life. GBS colonization may lead to late abortions, premature delivery, or stillbirth and in the newborn child, may result in sepsis, pneumonia or meningitis, all of which carry a significant risk of severe morbidity, long- term disability or death. With no general implemented and fully protective preventative treatment available for GBS, this underlines the unmet medical need of developing and providing a vaccine to prevent adverse pregnancy outcomes and life- threatening infections in infants caused by GBS. Furthermore, older adults and adults with certain co-morbidities such as diabetes or obesity are also at an increased risk of severe GBS infections and form a second population which would benefit from a prophylactic vaccine against this potentially fatal disease.
MinervaX’s lead vaccine candidate, is a novel protein-only vaccine, based on fusions of highly immunogenic and proactive protein domains from selected surface proteins of GBS. The company is strongly committed and dedicated to advancing its novel vaccine and has successfully completed two Phase II clinical trials of its maternal vaccine against GBS and is in preparation to commence Phase III clinical trials in this indication. Data from MinervaX’s GBS vaccine is very positive, demonstrating an acceptable safety profile in pregnant people and their infants, with high immunogenicity, leading to functionally active antibodies, with the potential for broad coverage and protection, alleviating the need for excessive use of antibiotics.
Wacker Biotech will manufacture the active ingredients of MinervaX’s novel vaccine candidate, alongside performing the technology transfer, process validation and process characterization activities for later commercial manufacturing. Subsequently, Wacker Biotech will perform all key functions critical to ensure stable commercial supply at its site in Amsterdam, The Netherlands.
Per Fischer, CEO of MinervaX, said: “GBS can be life-threatening for newborn babies and is linked to over half a million preterm births annually. Following the EUR 54 million financing last year, our team is advancing the development of our novel prophylactic vaccine against GBS for the benefit of all populations at risk, worldwide. Wacker Biotech is a robust manufacturing partner with a strong track record in late clinical and commercial supply and we look forward to collaborating with the team ahead of commencing Phase III studies.”
Ronald Eulenberger, Managing Director of Wacker Biotech B.V. in Amsterdam, stated: “With our strong background in E. coli processes, process characterization, and process validation experiences, we at Wacker Biotech are perfectly suited to support MinervaX with its ongoing program for the prevention of invasive GBS disease.”
Details of MinervaX’s completed Phase II clinical trials in pregnant people can be found at https://clinicaltrials.gov/ under the identifiers NCT04596878 and NCT05154578. In addition to pregnant people, MinervaX is also pursuing Phase I development of its novel GBS vaccine in older adults, under identifier NCT05782179.
ENDS
For further information please contact:
MinervaX
Per Fischer | Chief Executive Officer
Email: info@minervax.com
Optimum Strategic Communications
Mary Clark / Zoe Bolt / Vareen Outhonesack
Email: minervax@optimumcomms.com
Tel: +44 (0) 203 882 9621
Wacker Chemie AG
Dr. Karsten Werth
Email: karsten.werth@wacker.com
Tel: +49 89 (0) 6279-1573
Notes to Editors:
About MinervaX
MinervaX is a Danish biotechnology company, established in 2010 to develop a prophylactic vaccine against Group B Streptococcus (GBS), based on research from Lund University. MinervaX is developing a GBS vaccine for maternal immunization, and also for vaccination of older/at risk adults. Phase II clinical data from its maternal vaccination program suggest a high efficacy, based on the preliminary Correlate of Protection data from a natural history study. MinervaX’s GBS vaccine is a protein-only vaccine based on fusions of highly immunogenic and protective protein domains from selected surface proteins of GBS, the Alpha-like protein family (AIpN). Given the broad distribution of proteins contained in the vaccine on GBS strains globally, it is expected that MinervaX’s GBS vaccine will confer protection against virtually 100% of all GBS isolates. www.minervax.com
About Wacker Biotech
Wacker Biotech is a full-service contract manufacturer of therapeutic proteins, live biopharmaceutical products (LBPs), plasmid DNA (pDNA), messenger ribonucleic acid (mRNA) and vaccines based on microbial systems. Wacker Biotech’s portfolio extends from strain/process development and analytical testing through to production for clinical and commercial applications. Wacker Biotech operates three GMP-compliant, FDA- and EMA-certified production plants at its Jena and Halle sites in Germany and in Amsterdam in the Netherlands. In addition, Wacker Biotech has had a plant in San Diego (Wacker Biotech US Inc.) since February 2021. Wacker Biotech GmbH, Wacker Biotech B.V. and Wacker Biotech US Inc. are wholly owned subsidiaries of Munich-based Wacker Chemie AG.
MinervaX ApS, Nordre Fasanvej 215, DK-2000 Frederiksberg, Denmark VAT no. DK32673287
- QRL-101 aims to reduce hyperexcitability-induced neurodegeneration, which is present in approximately 50 percent of all ALS patients
- Completed Phase 1 single-ascending dose (SAD) clinical trial of QRL-101 enrolled 88 participants; no reported significant safety concerns or serious adverse events
- MAD data expected to be reported in first half of 2025; results will support larger global studies in people living with ALS
CAMBRIDGE, Mass., September 10, 2024 – QurAlis Corporation (“QurAlis”), a clinical-stage biotechnology company driving scientific breakthroughs into powerful precision medicines that have the potential to alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative and neurological diseases, today announced that the company recently completed dosing of the first participant cohort in the Phase 1 multiple-ascending dose (MAD) clinical trial evaluating QRL-101 (QRL-101-03; NCT06532396). QRL-101 is a first-in-class selective Kv7.2/7.3 ion channel opener for the treatment of hyperexcitability-induced disease progression in ALS. Kv7.2 is a mis-spliced protein in sporadic ALS patients.
QRL-101-03 is a randomized, double-blind, placebo-controlled, single-site Phase 1 clinical trial designed to evaluate the safety, tolerability, and pharmacokinetics of multiple-ascending doses of QRL-101 in adult healthy volunteers. The study is expected to enroll approximately 60 participants, who will be randomized in a 9:3 ratio of QRL-101 to placebo into five planned cohorts. The dose range of QRL-101 for this MAD study was determined by results from QurAlis’ Phase 1 single-ascending dose (SAD) clinical trial (QRL-101- 01; NCT05667779). Of the 88 healthy participants in the SAD clinical trial, no significant safety concerns or serious adverse events have been reported for QRL-101.
“We are excited to complete dosing of our first participant cohort in our Phase 1 MAD clinical trial of QRL-101. In the SAD study, QRL-101 was shown to be well tolerated, with no significant safety concerns or serious adverse events,” said Doug Williamson, M.D., chief medical officer of QurAlis. “ALS is a devastating, fatal neurodegenerative disease and there are currently no therapies that can significantly extend patients’ lives. QRL-101 has the potential to be a first-in-class effective therapy for ALS patients suffering from hyperexcitability-induced motor neuron degeneration. We look forward to advancing the clinical program for QRL-101 so QurAlis can bring much-needed therapies to people living with ALS.”
“Motor system hyperexcitability occurs in approximately 50 percent of all ALS patients and is linked to potassium channel dysfunction,” said Leonard H. van den Berg, M.D., Ph.D., professor of neurology and chair, TRICALS. “QRL-101, is a highly selective Kv7.2/7.3 ion channel opener, which in preclinical models shows a strong potential to control motor neuron hyperexcitability-induced neurodegeneration with an attractive side effect profile. We are encouraged by the findings from the SAD study of QRL-101 and look forward to results from the MAD study.”
QurAlis anticipates reporting topline data from the Phase 1 MAD clinical trial of QRL-101 in the first half of 2025.
More information about the QRL-101 Phase 1 clinical trials can be found at www.clinicaltrials.gov.
About Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease, is a progressive neurodegenerative disease impacting nerve cells in the brain and spinal cord, reducing muscle function and control. ALS can be traced to mutations in more than 25 different genes and is often caused by a combination of multiple sub-forms of the condition. Cases usually cannot be predicted, although a small percentage are inherited. ALS has a devastating impact on patients and families. ALS patients’ average life expectancy is three years; after diagnosis patients only have two years to live. There is currently no cure for the disease.
About Kv7
Kv7.2/7.3 is a voltage-gated potassium channel whose role is crucial for the regulation of neuronal excitability and membrane potential. Kv7.2 is mis-spliced in sporadic ALS leading to loss of function and abnormal electrical activity in the spinal cord and brain. The activation of this channel shows the potential to decrease spinal and cortical/motor neuron excitability and to positively affect CMAP (compound muscle action potential). This suggests that this may be an effective therapeutic approach for ALS patients suffering from hyperexcitability-induced motor neuron degeneration.
About QurAlis Corporation
At QurAlis, we are neuro pioneers on a quest to cure. We work with a relentless pursuit of knowledge, a precise attention to craft, and an optimistic mindset to discover and develop effective precision medicines that have the potential to alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative and neurological diseases. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a robust precision medicine pipeline with therapeutic candidates aimed at modifying severe disease pathology in defined patient populations based on both disease-causing genetic mutation(s) and clinical biomarkers. For more information, please visit www.quralis.com or follow us on X @QurAlisCo or LinkedIn.
Contact:
Kathy Vincent
kathy.vincent@quralis.com
310-403-8951
- CD3 bispecific antibody developed to redirect T cell-mediated immunity toward B7-H7 expressing tumors, expanding monotherapy treatment possibility for a new patient population
- Company’s precision immunotherapy approach enables prospective identification of patients most likely to benefit from NPX372 with a defined clinical biomarker
Cambridge, MA –September 5, 2024 – NextPoint Therapeutics, a clinical-stage biotechnology company developing a new class of precision immuno-oncology and tumor-directed therapeutics targeting the novel B7-H7 axis, today has unveiled NPX372, a novel T cell engager. NPX372 further expands NextPoint’s multi-modal focus on the emerging B7-H7 axis in cancer therapy.
B7-H7, also known as HHLA2, is an emerging immunomodulatory receptor upregulated in various solid tumor types, including colorectal carcinoma, non-small cell lung cancer, renal cell carcinoma, prostate cancer, and many others. Notably, this receptor is induced independently of PD-L1 or other B7 family members. Unlike other B7 family proteins that are expressed across a wide range of cell types, B7-H7 is primarily found on the epithelial cells of tumors, making it a unique and potentially specific target for tumor-directed therapies.
“T cell engagers have shown immense potential, but to date their application in solid tumors has remained a formidable challenge. NPX372 represents a significant advancement, aiming to redirect T cell-mediated immunity with high specificity by modulating key biological components, potentially offering more effective monotherapy treatment options for a range of solid tumors,” said Tatiana Novobrantseva, PhD, Chief Scientific Officer of NextPoint Therapeutics.
NPX372 is a CD3 bispecific antibody with unique capabilities to redirect T cell-mediated cytotoxicity toward B7-H7-positive tumors. In addition to CD3 engagement, this antibody interacts with the B7-H7 immune axis to achieve added potency. Preclinical data highlight NPX372’s potent anti-tumor responses and a favorable safety profile at clinically relevant doses with no indication of cytokine release syndrome. This asset is part of NextPoint's diverse portfolio of immunotherapies designed to target various tumor types. NextPoint is rapidly advancing the Investigational New Drug (IND) application for NPX372.
“NPX372 represents a significant advancement in our pursuit of precision immunotherapy," said Ivan Cheung, CEO of NextPoint Therapeutics. “As part of our ongoing immune checkpoint clinical programs, NPX267 and NPX887, we have developed a clinical biomarker for B7-H7 expression, which allows us to selectively target patients across various tumor types who may benefit from a potent T cell engager such as NPX372. This precision medicine approach allows us to potentially address solid tumors expressing B7-H7, tailoring treatments to those who will respond best. Our deep knowledge of B7-H7 biology drives our leadership in advancing innovative, transformative treatments that can make a meaningful difference in the lives of cancer patients.”
About NextPoint Therapeutics
NextPoint is launching a new world of precision immuno-oncology and tumor-targeting therapeutics through its leading scientific work on the novel B7-H7/HHLA2 axis. Our innovative approach integrates foundational science with a defined clinical biomarker to identify the right patient population for each B7-H7-directed therapy, so that we can deliver a new class of monotherapies for patients. Our team of proven drug developers is simultaneously advancing therapeutic approaches blocking the B7-H7 immune signaling pathway and utilizing the unique upregulation of B7-H7 in cancer as an anchor for tumor-targeting treatment modalities. To learn more, visit nextpointtx.com.
Contacts
Media Contact
Lauren Arnold
LA Communications
Lauren@lacommunications.net
NodThera Named a ‘Fierce 15’ Company by Fierce Biotech Philadelphia, PA, August 5, 2024 - NodThera, a leading clinical-stage biotech delivering a paradigm shift in the treatment of chronic inflammatory diseases through selective modulation of the NLRP3 inflammasome, today announces that it has been named by Fierce Biotech as one of 2024’s ‘Fierce 15’ biotechnology companies, designating it as one of the most innovative and promising biotechnology companies in the industry.
Daniel Swisher, Chief Executive Officer of NodThera, commented: “Over the past year, NodThera has made significant progress towards realizing the full potential of targeting the NLRP3 inflammasome, supported by a proven leadership team, best-in-class molecules and compelling pre-clinical and clinical data. We are incredibly honored to be profiled by Fierce Biotech among the industry’s most exciting and innovative companies. With preparations underway for our Phase II clinical development program in cardiovascular disease, obesity, and neuro-inflammation among other value inflection points, this achievement underscores the strength of our science and commitment to leading a paradigm shift in the treatment of chronic inflammatory diseases.”
Ayla Ellison, Editor-in-Chief, Fierce Life Sciences and Healthcare, said: “For the past 22 years, we have evaluated hundreds of companies for inclusion in the ‘Fierce 15’ special report. Our selection process considers various factors, including technological robustness, strategic partnerships, venture support and market positioning. This report highlights innovation and creativity amid intense competition.”
This year’s full list of winners can be viewed here: https://www.fiercebiotech.com/special- reports/introducing-fierce-biotechs-2024-fierce-15
For more information about NodThera please contact:
NodThera
Tel: +44 (0) 1223 608130
Email: info@nodthera.com
ICR Consilium
Amber Fennell, David Daley, Sukaina Virji
Tel: +44 (0)20 3709 5700
Email: nodthera@consilium-comms.com
About NodThera
NodThera is a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases. Led by an experienced management team, NodThera is combining a deep understanding of NLRP3 inhibition, pharmaceutical neuroscience expertise and precision chemistry. Its two lead clinical candidates are oral, small molecule NLRP3 inflammasome inhibitors, which have demonstrated differentiated, potentially best-in-class clinical profiles with significant anti-inflammatory effects and high brain penetration, offering distinct opportunities to treat multiple indications. The Company is backed by top-tier investors including 5AM Ventures, Blue Owl Capital, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Philadelphia, Pennsylvania, with additional operations in Cambridge, UK and Seattle, WA. Learn more at www.nodthera.com or follow the Company on LinkedIn.
About Fierce Biotech
Fierce Biotech is the biotech industry’s daily monitor, providing the latest news, articles, and resources related to clinical trials, drug discovery, FDA approval, FDA regulation, patent news, pharma news, biotech company news and more. More than 300,000 top biotech professionals rely on Fierce Biotech for an insider briefing on the day’s top stories.
Greg brings over 25 years of public and private company leadership experience
Philadelphia, PA, August 1, 2024 - NodThera, a leading clinical-stage biotech delivering a paradigm shift in the treatment of chronic inflammatory diseases through selective modulation of the NLRP3 inflammasome, today announces the appointment of Greg Chow as Chief Financial and Business Officer (CFBO), effective immediately.
Greg is an experienced executive with over 25 years of corporate finance, capital markets, investment banking, financial accounting, drug development operations, and business development experience. Over the course of his career, he has led major financing rounds for both public and private companies, as well as supported operations and administrative functions.
Greg joins NodThera from Freenome Holdings, where he served as Chief Financial Officer (CFO) and helped successfully execute the company’s recent $254 million Series F financing round. Prior to Freenome, Greg was CFO at Frontier Medicines, where he led its Series B and C fundraising rounds and headed up Alliance Management for its global, multi-year collaboration with Abbvie. His leadership across Frontier’s business operations was instrumental in helping the company transition from a discovery-stage to clinical-stage organization. Greg gained significant public company experience as Executive Vice President and CFO at Aptose Biosciences (Nasdaq: APTO), overseeing the company’s dual listing on the Nasdaq and Toronto stock exchanges in addition to successfully raising over $225 million and attracting multiple new shareholders.
Prior to his time as an industry executive, Greg spent 14 years in investment banking as Managing Director and Director of Private Placements at Wedbush Securities and in senior roles at RBC Capital Markets and Wells Fargo Securities. Greg holds an MBA in Finance from the Wharton School at the University of Pennsylvania and a BA in Business Economics from the University of California, Santa Barbara. He is a Certified Public Accountant (inactive) in the state of California.
Daniel Swisher, Chief Executive Officer of NodThera, said: “Greg’s financial experience and extensive track record of long-term value creation for both private and public biotechs will be invaluable as NodThera continues to execute its strategy and evolves into a mature, high-value, clinical-stage company. I look forward to working closely with him as part of our executive team. Greg will be a key strategic partner as we continue to unlock the broad potential and maximum value of our highly differentiated brain-penetrant NLRP3 inflammasome inhibitors in the treatment of chronic inflammatory diseases. On behalf of the whole team, I would like to wish him a very warm welcome to NodThera.”
Greg Chow, Chief Financial Officer of NodThera, added: “I am incredibly excited to join NodThera at such a pivotal time in its development. The potential to modulate inflammation for the treatment of chronic diseases presents a significant opportunity to impact patients’ lives in a meaningful way. I look forward to being immersed in one of the most exciting and rapidly evolving therapeutic categories in biotech today. With a highly accomplished team, positive progression of lead candidate NT-0796 and upcoming Phase II studies in obesity, cardiovascular and Parkinson’s disease, NodThera is well positioned in its current trajectory with some important inflection points on the near-term horizon. I
am eager to bring my capital markets, operations, and business development experience to NodThera and look forward to partnering with Dan, the Board of Directors, and the wider team.”
For more information about NodThera please contact:
NodThera
Tel: +44 (0) 1223 608130
Email: info@nodthera.com
ICR Consilium
Amber Fennell, David Daley, Sukaina Virji
Tel: +44 (0)20 3709 5700
Email: nodthera@consilium-comms.com
About NodThera
NodThera is a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases. Led by an experienced management team, NodThera is combining a deep understanding of NLRP3 inhibition, pharmaceutical neuroscience expertise and precision molecular chemistry. Its two lead clinical candidates are oral, small molecule NLRP3 inflammasome inhibitors, which have demonstrated differentiated, potentially best-in-class clinical profiles with significant anti-inflammatory effects and the ability to penetrate different areas of the brain, offering distinct opportunities to treat multiple indications. The Company is backed by top-tier investors including 5AM Ventures, Blue Owl Capital, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Philadelphia, Pennsylvania, with additional operations in Cambridge, UK and Seattle, WA. Learn more at www.nodthera.com or follow the Company on LinkedIn.
Paris, July 10, 2024 — In a groundbreaking development, Eligo Bioscience has made in vivo bacterial genome editing a reality. In work published in the journal Nature, Eligo demonstrates, for the first time, the ability to precisely and efficiently edit the genome of bacteria directly in the gut. Achieving gene editing with nearly 100% efficiency and without disturbing the surrounding microbial communities is a major milestone in microbial gene therapy, paving the way for the development of new treatments for microbiome-associated diseases.
A novel chapter for gene editing: from human genes to bacterial genes
Since the advent of gene therapies in the 1970s, culminating in the recent approval of the first CRISPR-based drugs, researchers have made significant strides in delivering DNA to various cell types and organs to correct genetic defects. Until now, however, a large proportion of the genes carried by humans remained completely out of reach: the microbiome.
The microbiome, composed of billions of commensal bacteria, is essential to our health and the proper functioning of our immune system. However, we are discovering a growing number of bacterial genes implicated in chronic and serious diseases: from bacterial peptides that can trigger autoimmune diseases to bacterial virulence factors that contribute to inflammatory diseases, tumor formation, neurodegenerative diseases. As it becomes clearer that conventional therapeutics affecting microbiome composition can cause serious adverse events and side effects, it is critical to develop approaches to edit the microbiome in a targeted way.
"What Eligo Bioscience has achieved shows that it’s now possible to make specific changes to the DNA of bacteria in the gut, similar to how scientists have been editing human genes to investigate or treat genetic disorders," said David Bikard, co-founder of Eligo Bioscience and researcher at the Institut Pasteur in Paris.
A precise and efficient method
Achieving efficient in vivo gene editing of bacteria requires both an effective delivery method and a robust editing system.
In a ‘tour de force,’ Eligo demonstrated the ability to engineer, produce, and purify a bacteriophage-derived capsid containing a synthetic DNA payload encoding a base-editor system. After oral administration to mice, the capsid delivered the payload with extreme efficiency and precision to target bacterial populations residing among the hundreds of bacterial species in the mouse gut. Remarkably, the system could precisely inactivate antibiotic resistance genes or virulence factors by creating single-base pair mutations in the corresponding genes. When targeting E. coli strains colonizing the mouse gut, the technology modified the target gene in over 90% of the bacteria, reaching up to 99.7% in some cases.
These modifications remained stable for at least 42 days.
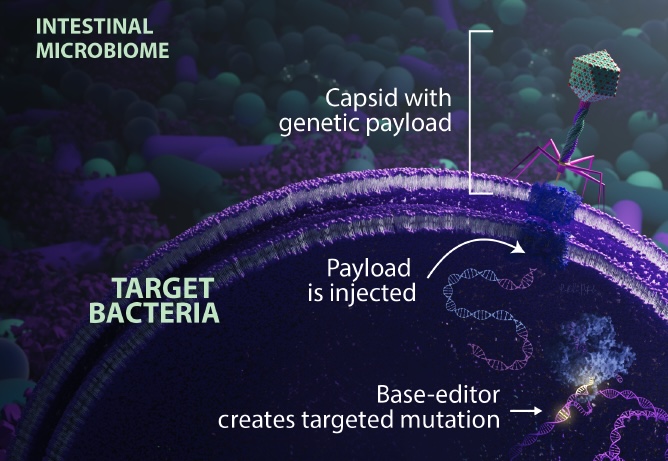
Jesus Fernandez, Eligo VP of Technology and a senior author of the study, highlights how "this achievement is the culmination of eight years of work by the team at Eligo Bioscience and represents a paradigm shift in microbiome research. This leap forward provides Eligo with a unique edge to develop microbial genetic medicines, and also to find novel therapeutic targets with a novel tool to interrogate the role and function of bacterial genes in health and disease."
Looking Ahead
Eligo Bioscience is already using this novel and patented approach to create new treatments for various conditions that affect millions of people worldwide. "This technology enhances Eligo's pioneering arsenal of in-vivo editing tools, and can be applied to various bacteria and genes, opening the door to treatments for a wide range of health issues", said Xavier Duportet, CEO and co-founder of Eligo. ‘It therefore broadens the landscape of addressable therapeutic targets in the gene editing field’.
Read the paper in Nature here.
–
About Eligo Bioscience
Eligo Bioscience is the world leader in microbiome in-vivo gene editing and is advancing a highly differentiated pipeline of precision medicines to address unmet medical needs driven by the expression of deleterious bacterial genes in immuno-inflammation, oncology, and infectious diseases.
Eligo’s first drug candidates advancing to the clinic are based on the delivery of CRISPR-Cas systems to kill bacteria based on their genetic signature. One of these programs targets
pro-inflammatory genes in skin bacteria driving moderate-to-severe acne pathology in dozens of millions of patients around the world. This lead program is expected to enter clinical trials in patients in 2025.
Eligo was named a Technology Pioneer by the World Economic Forum and has raised over €50M from top-tier investors including Sanofi Ventures, Khosla Ventures, BPIFrance, and Seventure Partners.
Eligo was founded by Luciano Marraffini (Professor at The Rockefeller University and cofounder of Intellia Therapeutics, Timothy Lu (Professor at MIT, and CEO at Senti Biosciences), Dr. David Bikard (Professor at Institut Pasteur) and Dr. Xavier Duportet (MIT TR35, Young Global Leader, and Termeer Fellow).
For more information about Eligo Bioscience and our latest research, please visit Eligo’s website: https://eligo.bio/
Contact:
xavier.duportet@eligo-bioscience.com
- Company initiates partnership and collaboration with UMass Chan Medical School to characterize relevant targets that enable ASO-mediated correction for FXS
the leading inherited form of intellectual disability and a known cause of autism - Fragile X syndrome is a rare, genetic neurological disease that is
CAMBRIDGE, Mass., June 25, 2024 – QurAlis Corporation (“QurAlis”), a clinical-stage biotechnology company driving scientific breakthroughs into powerful precision medicines that have the potential to alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative and neurological diseases, today announced it is expanding its industry-leading precision medicine antisense oligonucleotide (ASO) splicing expertise beyond neurodegeneration into Fragile X syndrome (FXS).
As part of this strategy, QurAlis has initiated a partnership and collaboration with UMass Chan Medical School to explore the biology of FXS to determine and confirm relevant targets that could enable ASO- mediated correction for FXS. QurAlis can leverage its deep understanding, knowledge and expertise in developing ASOs including its proprietary FlexASO™ Splice Modulator Platform as part of the collaboration.
“QurAlis’ deep knowledge and expertise of ASO splicing targets combined with our FlexASO™ Splice Modulator Platform gives us the latitude to go beyond neurodegeneration to explore the potential of bringing much-needed precision medicines for other serious neurological diseases like FXS,” said Kasper Roet, Ph.D., chief executive officer and co-founder of QurAlis. “Our partnership and collaboration with UMass Chan in FXS underscores the enormous value of partnerships between academia and industry to drive innovation forward to help fulfill unmet medical needs for patients. We are excited to expand our scope into a new therapeutic area and to make use of our expertise in splicing targets so that QurAlis can continue the goal of making a real difference in patients’ lives.”
“We are excited to be partnering with QurAlis in the next step of this years-long research,” said Joel Richter, Ph.D., the Arthur F. Koskinas Chair in Neuroscience and professor of molecular medicine at UMass Chan Medical School in Worcester, MA. “QurAlis’ translational knowledge and clinical trial experience in ASO splicing for neurodegenerative disorders is an important component to bridging the gap between biomedical discoveries made in the laboratory and delivering therapies to patients in the clinic. With their help, we are hopeful that one day we may be able to offer patients with FXS meaningful treatment options.”
About Fragile X Syndrome (FXS)
Fragile X syndrome is the leading inherited form of intellectual disability and a known cause of autism. It is a genetic condition caused by a mutation of a single gene – FMR1 – on the X chromosome. This mutation of FMR1 causes a range of developmental problems including learning disabilities, behavioral challenges, and cognitive impairment.
An orphan disease, FXS affects approximately 87,000 individuals in the U.S. alone – one in 4,000 men and one in 6,000 women. Though FXS occurs in both genders, males are more frequently affected than females, and generally with greater severity. In addition to intellectual disability, FXS patients endure a wide range of disabling symptoms including severe anxiety, social aversion, hyperactivity and attention deficit, sensory hypersensitivity, aggression, developmental seizures, and others. There are no disease- modifying therapies currently available for FXS.
About QurAlis’ FlexASOTM Splice Modulator Platform
Antisense oligonucleotides (ASOs) are short, engineered single-stranded DNA/RNA molecules that can selectively bind RNA to regulate its expression in the cell. ASO technology has been leading in the field of protein regulation and has since allowed us to develop treatments for neurodegenerative disease by changing the expression of genes connected to the disease.
QurAlis’ FlexASOTM Splice Modulator Platform was developed to generate splice-switching ASOs with improved potency, increased therapeutic index and improved bio-distribution. This bespoke platform has the potential to tackle the spectrum of neurodegenerative and neurological diseases.
About QurAlis Corporation
At QurAlis, we are neuro pioneers on a quest to cure. We work with a relentless pursuit of knowledge, a precise attention to craft, and an optimistic mindset to discover and develop effective precision medicines that have the potential to alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative and neurological diseases. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a robust precision medicine pipeline with therapeutic candidates aimed at modifying severe disease pathology in defined patient populations based on both disease-causing genetic mutation(s) and clinical biomarkers. For more information, please visit www.quralis.com or follow us on X @QurAlisCo or LinkedIn.
Contact:
Kathy Vincent
kathy.vincent@quralis.com
310-403-8951
Industry veteran, founder of Medivation and serial entrepreneur joins T-Therapeutics to support development of the company’s pipeline of TCR-based immuno-oncology drugs
12 June 2024; Cambridge, England – T-Therapeutics, a biotechnology company developing next- generation soluble TCR therapeutics targeting cancer and autoimmune indications, today announces the appointment of biopharma industry veteran Dr David Hung as Chairman of its Board of Directors.
Dr Hung is a hugely successful serial entrepreneur in the oncology space. He founded Medivation in 2003 which was sold to Pfizer for $14.3 billion in 2016. At Medivation, Dr Hung identified, in-licensed and led bench-to-bedside development of enzalutamide (XTANDI®) for advanced prostate cancer, taking it from first in vitro laboratory experiment to FDA approval in seven years, one of the fastest development timelines in pharmaceutical history. XTANDI, approved in more than 60 countries is one of the top cancer drugs by revenue, reaching blockbuster drug status and exceeding $6 billion in global annual sales in 2023.
Prior to founding Medivation, Dr Hung served as President and CEO of ProDuct Health, a venture- backed startup medical device company founded in 1998 that developed, manufactured and commercialized a breast microcatheter – invented by Dr Hung – for breast cancer risk assessment. ProDuct Health was acquired in 2001 for $168 million by Cytyc Corporation.
Dr Hung is currently President and CEO of Nuvation Bio, which he founded in 2018, a late-stage biopharma developing differentiated and novel therapeutic candidates to tackle some of the greatest unmet needs in oncology.
Dr Hung received an A.B. summa cum laude in biology from Harvard College and an M.D. Alpha Omega Alpha from the University of California, San Francisco, School of Medicine. He completed simultaneous clinical fellowships in hematology, oncology and transfusion medicine as well as two basic science research fellowships in molecular biology at the University of California, San Francisco, School of Medicine.
Professor Allan Bradley, CEO of T-Therapeutics, commented: “Having followed David’s career for many years, I’m delighted that he is joining T-Therapeutics’ Board as Chair. David brings a wealth of scientific, clinical and company building experience to our Board. I look forward to drawing on his deep understanding of drug development in the oncology space, as well as his substantial experience in building biotechnology companies as we work to develop a pipeline of drugs designed to reshape the clinical landscape for cancer patients.”
Dr David Hung, Chairman of T-Therapeutics, also commented: “I’m excited to work with Allan and his very experienced team to help realise their vision. T-Therapeutics has an unusually strong and unique scientific foundation that is being leveraged to build a pipeline of transformative medicines. The team have an excellent track record of successful development of drugs that leverage immune cell biology. I believe their unique TCR platform has the potential to deliver a pipeline of transformative medicines.”
In November 2023 T-Therapeutics announced a £48m series A fundraise for the development of next-generation TCR therapeutics to transform cancer treatment, led by Sofinnova Partners, F-Prime Capital, Digitalis Ventures and Cambridge Innovation Capital with participation from Sanofi Ventures and the University of Cambridge Venture Fund.
– ENDS –
Contact Us
T-Therapeutics
Allan Bradley
info@t-therapeutics.com
ICR Consilium
Amber Fennell, Sukaina Virji
t-therapeutics@consilium-comms.com
About T-Therapeutics
T-Therapeutics is a next-generation T cell receptor (TCR) company spun off from the University of Cambridge. The company was created to harness the power of T cell biology, evolved over millions of years, to create safe and effective treatments for many cancers and autoimmune diseases. T- Therapeutics combines world-leading expertise in mouse genome engineering, deep knowledge and experience in biopharmaceutical drug development, single cell genomics, machine-learning and structural biology, anchored in a culture of creativity and collaboration. T-Therapeutics is developing ‘optimal’ TCR based therapeutics using a proprietary OpTiMus® platform, based on a fully humanized TCR mouse that provides an almost unlimited source of unique, antigen-specific human TCRs. These TCRs are directed at multiple target classes, many of which have never been worked on before. The company is developing a pipeline of first-in-class drugs that are intended to become transformative medicines, reshaping the clinical landscape for patients with cancer or autoimmune diseases.
- Brain-penetrant NLRP3 inflammasome inhibitor demonstrates highly significant and rapid reduction of key inflammatory marker CRP compared to placebo
- Reductions in multiple markers of cardiometabolic risk confirm profound antiinflammatory effect of NT-0796
- Most pronounced weight loss seen among high-risk subgroups
- Planning is underway for Phase II studies in obesity, Parkinson’s disease and other cardiometabolic indications
BOSTON, MA, June 12, 2024 - NodThera, a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases, today announces positive data from its Phase Ib/IIa cardiovascular risk study in inflamed obese subjects, evaluating the effects of its oral, brain-penetrant NLRP3 inflammasome inhibitor NT-0796, on inflammatory, cardiovascular and metabolic risk parameters.
Obese subjects with elevated baseline C-reactive protein (CRP), measured using an hsCRP assay, were recruited. Elevated CRP is a key marker of chronic inflammatory diseases such as coronary artery disease, and one or more cardiovascular risk factors (e.g. metabolic syndrome, prediabetes, diabetes, hyperlipidemia or hypertension). The study was conducted in-clinic for 28 days and energy intake was limited to 2000 kCal per day. At study end, a highly statistically significant and rapid CRP reduction in NT-0796 dosed subjects was observed compared to placebo, meeting the primary endpoint of the study. Furthermore, more than 75% of NT-0796 dosed subjects achieved a CRP reduction at day 28 to ≤2mg/L compared to less than 25% among the placebo group, a value generally regarded as a threshold for reducing cardiovascular risk.
NodThera’s study also demonstrated reductions in additional pro-inflammatory and cardiometabolic biomarkers, secondary endpoints that build on the previously reported diet-induced obesity preclinical study and Phase Ib/IIa Parkinson’s disease clinical study. Key reductions in cardiovascular and metabolic biomarkers were found to be highly translatable and consistent with the importance of targeting both brain and peripheral NLRP3. While all subjects lost weight due to calorie restriction in both the active and placebo groups, the most pronounced placebo-adjusted reductions in body weight were among high-risk subgroups of NT-0796 dosed subjects, and, with the Company’s earlier preclinical data, support the anti-obesity potential of the molecule.
Preparations are underway for a Phase II study in obesity and other Phase II studies including additional cardiometabolic diseases and Parkinson’s disease.
Alan Watt, President & Chief Scientific Officer of NodThera, said: “This is an impressive data set, made all the more remarkable by being achieved within 28 days. In addition to the profound anti- neuroinflammatory effects in Parkinson’s disease patients demonstrated previously, the data generated in this study of NT-0796 show improvements in inflammatory and metabolic biomarkers of CV risk that go beyond that seen with antibody and peptide therapies. With additional reductions in body weight that should be enhanced with longer-duration dosing, NT-0796 is confirmed as a highly promising therapeutic agent for chronic inflammatory indications.”
As before, NT-0796 was generally safe and well tolerated; adverse events (AEs) were mainly mild and transient, and no serious adverse events (SAEs) were observed.
Taken together, the findings demonstrate that NT-0796 successfully delivered anti-inflammatory changes in an obese inflamed population, amelioration of cardiometabolic risk factors and reductions in body weight within 28 days, indicating excellent potential for long-term, oral dosing in obese subjects.
For more information about NodThera please contact:
NodThera
Tel: +44 (0) 1223 608130
Email: info@nodthera.com
ICR Consilium
Amber Fennell, David Daley, Sukaina Virji
Tel: +44 (0)20 3709 5700
Email: nodthera@consilium-comms.com
About NodThera
NodThera is a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases. Led by an experienced management team, NodThera is combining a deep understanding of NLRP3 inhibition, pharmaceutical neuroscience expertise and precision molecular chemistry. Its two lead clinical candidates are oral, small molecule NLRP3 inflammasome inhibitors, which have demonstrated differentiated, potentially best-in-class clinical profiles with significant anti-inflammatory effects and the ability to penetrate different areas of the brain, offering distinct opportunities to treat multiple indications. The Company is backed by top-tier investors including 5AM Ventures, Blue Owl Capital, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Boston, MA, with additional operations in Cambridge, UK and Seattle, WA. Learn more at www.nodthera.com or follow the Company on LinkedIn.
- Industry veteran Williamson brings nearly three decades’ experience as a leader in neuroscience R&D at organizations including Eli Lilly and Company, Lundbeck, Parexel, and Acadi Pharmaceuticals
- Former Karuna CFO Brown brings more than 15 years of corporate finance, strategy, business development, M&A, and company-building experience in the biopharmaceutical industry
- Highly respected biotech executive Virani brings deep experience in corporate development, life science R&D, and building successful companies
CAMBRIDGE, Mass., June 11, 2024 – QurAlis Corporation (“QurAlis”), a clinical-stage biotechnology company driving scientific breakthroughs into powerful precision medicines that will alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative diseases, today announced that it has appointed Douglas (Doug) Williamson, M.D., as chief medical officer (CMO) and Jason Brown, MBA, as chief financial officer (CFO). Dr. Williamson succeeds Angela Genge, M.D., who continues in a consulting role for QurAlis. The company also announced the appointment of Shafique Virani, M.D., to its board of directors.
“Doug is an expert drug developer for neurological disorders, talented pharmaceutical executive, and dedicated physician who brings a breadth of successful experience across all phases of clinical development – from the interface of discovery with first-in-human trials, through Phase 2 dose-range finding studies, Phase 3 pivotal trials, and post-market lifecycle optimization,” said Kasper Roet, Ph.D., chief executive officer (CEO) and co-founder of QurAlis.
Dr. Roet continued, “Jason’s experience and track record in raising capital, preparing companies to go public, scaling companies, and executing partnership and M&A deals, will be instrumental to QurAlis’ next phase of growth. Jason’s outstanding profile in finance and operational growth combined with Doug’s extensive global R&D experience, will greatly complement our team as we execute on our strategy to bring promising new precision medicines to patients with ALS, FTD, and other neurodegenerative and serious neurological diseases rapidly.”
“Neurodegenerative diseases like ALS and FTD are areas of huge unmet medical need, and QurAlis is a company that is changing the future of how we treat these diseases by pioneering the development of powerful precision medicines,” said Dr. Williamson. “QurAlis’ cutting-edge science and Kasper’s leadership is a powerful combination. I am thrilled to join such a dedicated team and apply my clinical and drug development experience toward building this innovative biopharma company to bring new, life- changing treatment options to patients with serious diseases and making a real difference in patients’ lives.”
“QurAlis is a mission-driven organization dedicated to bringing transformative therapies to patients with ALS and other serious neurodegenerative diseases,” said Mr. Brown. “This is an exciting time for QurAlis and I am thrilled to join QurAlis and work closely with Kasper, our board, and the broader team to advance our corporate strategy and lead the company’s financial activities as we further scale our organization and rapidly advance clinical development to bring much-needed medicines to patients.”
Dr. Williamson has almost thirty years of leadership experience in all phases of neuroscience clinical drug development and has led several successful investigational new drug (IND) applications, global new drug applications (NDAs), and commercial launches in a broad range of psychiatry and neurology indications. He spent many years in senior leadership positions at large biopharma organizations including Eli Lilly and Company, Parexel International, and Lundbeck, where he led research and development (R&D) in the U.S. More recently, Dr. Williamson has gained experience at smaller biotechnology companies including as CMO at Avadel Pharmaceuticals, and head of R&D at Acadia Pharmaceuticals. He has consulted for numerous companies on neuroscience clinical development strategy. He graduated in medicine from Edinburgh University in Scotland and held a full-time academic appointment at Oxford University prior to joining the industry. Dr. Williamson is a member of the Royal College of Psychiatrists and the National Academy of Medicine’s Neuroscience Forum.
Before joining QurAlis, Mr. Brown was the CFO of Karuna Therapeutics. He joined Karuna in 2018 as vice president (VP) of corporate finance and its first finance employee. Throughout his tenure at Karuna, Mr. Brown drove the growth of both the finance department and broader organization, which included supporting multiple equity financings, business development transactions, and ultimately the acquisition by Bristol-Myers Squibb for $14 billion in 2024. Previously, Mr. Brown worked at PureTech Health as VP of corporate finance where he was responsible for the financing and operational growth of the company. He also spent five years at Novartis in roles of increasing responsibility within the finance department. He holds an MBA from Boston College and BA from Hamilton College.
Dr. Roet continued, “On behalf of the team, I would like to thank Angela Genge for her leadership and many contributions to QurAlis and we are delighted that she continues to be an integral member of our team.”
Biopharma leader Shafique Virani, M.D., joins QurAlis’ board of directors
QurAlis also announced the appointment of Shafique (Shaf) Virani, M.D., to its board of directors.
Dr. Roet said, “I am delighted to welcome Shaf to our board of directors. His success as a biopharma leader has given hope to patients around the world, and his perspectives will help QurAlis build our capabilities, drive growth for our investors, and realize our vision of accelerating the development of life-changing medicines to help patients with serious neurodegenerative and neurological diseases.”
“Having spent most of my career in neuroscience, I am excited about QurAlis’ groundbreaking science and its robust pipeline and platforms,” said Dr. Virani. “QurAlis’ pioneering approach could revolutionize how we treat serious neurodegenerative diseases and offers tremendous hope to patients, particularly those with intractable diseases like ALS for which there remains significant unmet medical need. I am impressed by QurAlis’ leadership and look forward to working with Kasper and the leadership team and my fellow board members to grow the business and shareholder value and pursue QurAlis’ mission to improve patients’ lives.”
Dr. Virani currently serves as chief business officer at Noetik, Inc., an AI-native biotechnology company focused on discovering and developing cancer therapies. Before joining Noetik in March 2024, he served as Recursion’s chief business officer for four years, as well as interim chief medical officer for half that time. At Recursion, Dr. Virani was instrumental in forging transformative partnerships with Bayer and Roche/Genentech, the latter being one of the largest drug discovery partnerships ever consummated in industry. Previously, Dr. Virani was chief executive officer (CEO) of Navire Pharma and CoA Therapeutics, each a subsidiary of BridgeBio Pharma, Inc., where he also served as CEO-in-residence.
Prior to BridgeBio, Dr. Virani assumed a 13-year-long tenure at Genentech/Roche in several senior roles, most recently as vice president, business development, licensing M&A, and global head of neuroscience, rare disease, and ophthalmology partnering. There, he helped build a portfolio of medicines including Evrysdi® (risdiplam) for spinal muscular atrophy, Enspryng® (satralizumab-mwge) for neuromyelitis optica spectrum disorder, and several therapeutics in the mid-to-late-stage clinical pipeline via licensing and acquisitions. Dr. Virani trained as a neurosurgeon in Cambridge, UK and Boston and received his M.D. from the University of Nottingham, UK.
About QurAlis Corporation
At QurAlis, we are neuro pioneers on a quest to cure. We work with a relentless pursuit of knowledge, a precise attention to craft, and an optimistic mindset to discover and develop effective precision medicines that will alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative diseases. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a pipeline with therapeutic candidates that target specific components of ALS and FTD pathology and defined patient populations based on both disease-causing genetic mutation(s) and clinical biomarkers. For more information, please visit www.quralis.com or follow us on X @QurAlisCo or LinkedIn.
Contact:
Kathy Vincent
kathy.vincent@quralis.com
310-403-8951
- QRL-204 is a splice-switching ASO generated through QurAlis’ FlexASO™ Platform; represents Lilly’s first program targeting UNC13A in ALS and FTD
- Parties to also collaborate to leverage QurAlis’ ALS and ASO development expertise to advance QRL-204 and next-generation compounds
- UNC13A is an essential regulator of neurotransmitter release at synapses; mis-splicing is a critical RNA alteration occurring in up to 63 percent of all ALS patients and up to one-third of all FTD cases
CAMBRIDGE, Mass., June 3, 2024 – QurAlis Corporation (“QurAlis”) today announced that it has entered into an exclusive license agreement with Eli Lilly and Company (“Lilly”) in which QurAlis is granting Lilly global rights to develop and commercialize QRL-204, a potentially best-in-class splice-switching antisense oligonucleotide (ASO) designed to restore UNC13A function in amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative diseases.
Under the terms of the agreement, QurAlis granted Lilly an exclusive, worldwide license to develop and commercialize QRL-204 and other UNC13A-targeting compounds in exchange for an upfront payment of $45 million to QurAlis, plus an additional equity investment. QurAlis is also eligible for future milestone payments of up to $577 million and tiered royalties on net sales.
The agreement includes a research and development (R&D) collaboration to identify and develop additional candidates targeting UNC13A, leveraging QurAlis’ proprietary FlexASO™ Splice Modulator Platform. The QurAlis FlexASO Splice Modulator Platform was developed to generate splice-switching ASOs with improved potency and increased therapeutic index. QurAlis’ ASOs correct UNC13A mis-splicing, restore UNC13A protein production and reduce cryptic exons that may contribute to disease progression.
“We are determined at Lilly to uncover the next great idea, the next collaboration, the next advancement in technology that will drive us toward meaningful treatments for ALS and FTD patients. It’s all for one reason – to make life better for even more people,” said Andrew Adams, senior vice president, neurodegeneration research, and director, Lilly Institute for Genetic Medicine. “Genetic precision medicines like QRL-204 that target specific causal components of disease pathology hold great promise for delivering meaningful advances against a range of neurodegenerative diseases like ALS and FTD. We look forward to collaborating with QurAlis to identify and develop additional next-generation candidates targeting UNC13A.”
“On behalf of the entire team at QurAlis, we are extremely pleased to partner with Lilly, an innovation- led company advancing transformative medicines to help make life better for people around the world,” said Kasper Roet, chief executive officer and co-founder of QurAlis. “At QurAlis, we are driven to explore the deepest aspects of human neurology to find the treatments patients urgently need. This partnership enables QRL-204 to rapidly move toward the clinic, while we continue to advance our other lead programs. We look forward to combining our complementary strengths to uncover additional candidates that target UNC13A that have the potential to transform the treatment of neurodegenerative diseases like ALS and FTD and beyond.”
Amyotrophic lateral sclerosis is a progressive neurodegenerative disease characterized by the loss of neurons in the spinal cord, brainstem, and brain. A defining feature of both sporadic and familial disease is the cytoplasmic mis-localization of TAR DNA Binding Protein-43 (TDP-43). TDP-43 pathology is implicated in 90 percent of ALS cases and approximately 50 percent of FTD cases.
UNC13A is an essential regulator of neurotransmitter release at synapses and is one of several pre-mRNAs that becomes mis-spliced due to loss of nuclear TDP-43 in disease. Up to 63 percent of ALS patients and up to one-third of FTD patients carry a single nucleotide polymorphism in the UNC13A gene or show TDP- 43 pathology, which greatly exacerbates UNC13A mis-splicing leading to loss of function of the UNC13A protein.
Preclinical data recently presented at the AD/PD™ 2024 International Conference on Alzheimer’s and Parkinson’s Diseases and Related Neurological Disorders showed QurAlis’ UNC13A splice-switching ASOs modulate UNC13A splicing and restore normal synaptic activities in ALS and FTD. QurAlis’ ASOs prevented cryptic exon inclusion in UNC13A transcripts, increased UNC13A protein levels, and normalized localization of UNC13A protein at the synapse.
About QurAlis Corporation
At QurAlis, we are neuro pioneers on a quest to cure. We work with a relentless pursuit of knowledge, a precise attention to craft, and an optimistic mindset to discover and develop effective precision medicines that will alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative diseases. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a pipeline with therapeutic candidates that target specific components of ALS and FTD pathology and defined patient populations based on both disease-causing genetic mutation(s) and clinical biomarkers. For more information, please visit www.quralis.com or follow us on X @QurAlisCo or LinkedIn.
Contact:
Kathy Vincent
kathy.vincent@quralis.com
310-403-8951
- Dan joins NodThera with over 30 years of pharmaceutical industry leadership experience, including as President and COO of Jazz Pharmaceuticals and CEO of Sunesis Pharmaceuticals
- Interim CEO Alan Watt becomes President and CSO including leadership of R&D to further build out NodThera’s pioneering work in CNS modulation of chronic inflammatory diseases
BOSTON, MA, May 28, 2024 - NodThera, a leading clinical-stage biotech delivering a paradigm shift in the treatment of chronic inflammatory diseases through selective modulation of the NLRP3 inflammasome, today announces the appointment of Daniel Swisher as Chief Executive Officer (CEO), to strengthen the Company’s commercial capabilities, effective immediately. Alan Watt, the interim CEO moves to an expanded role as President and Chief Scientific Officer (CSO).
Dan joins NodThera with significant leadership expertise of over 30 years in the life sciences industry. During his career, he has led the advancement and commercialization of novel therapeutics across geographic regions and in multiple therapeutic areas, including neuroscience, oncology and rare diseases. He brings to NodThera significant private and public company experience driving cross functional, high-performing teams and building relationships with investors, analysts and corporate partners.
Dan joins NodThera from Jazz Pharmaceuticals (NASDAQ: JAZZ), where he recently served as President and Chief Operating Officer. At Jazz, he was instrumental in the geographic and therapeutic expansion of the company’s commercial and R&D pipeline, as well as playing a critical role in many key corporate development transactions. Prior to his time at Jazz, Dan was CEO and President of Sunesis Pharmaceuticals, guiding the evolution of its early-stage chemistry platform into a multi-asset development platform. During his CEO tenure at Sunesis, he oversaw equity capital market fundraises totaling over $400 million and led the Group to an IPO on the Nasdaq stock exchange.
Earlier in his career, Dan served as Senior Vice President of Sales and Marketing at ALZA Corporation, where he was responsible for its flagship pharmaceutical businesses based in the United States. He also played a pivotal role in the company’s growth into a fully integrated specialty pharmaceutical company and eventual $10.5 billion acquisition by Johnson & Johnson.
Alan Watt, who as the Company’s founder has also served as NodThera’s interim CEO, becomes President and CSO. He will be responsible for the growing R&D organization and focused on strengthening NodThera’s world-leading position in NLRP3 inflammasome inhibition research, with a focus on modulation of neuroinflammation for the treatment of obesity, cardiometabolic disease and neurodegeneration.
Don Nicholson, Executive Chair of the Board of Directors of NodThera, said: “Dan is an accomplished, results-driven leader with a proven track record in developing and commercializing novel therapeutics and building strong, successful businesses. We look forward to drawing on his deep understanding of the biopharmaceutical and financial markets, as well as his substantial experience in advancing clinical-stage assets towards commercialization, as we progress our broad pipeline of NLRP3 inflammasome inhibitors to deliver a paradigm shift in the treatment of chronic inflammatory diseases.
“We are extremely grateful for Alan’s strategic and scientific leadership during his tenure as interim CEO. His creativity, determination and integrity have been invaluable to NodThera in shaping the Company’s strategy and we are delighted that NodThera will continue to benefit from his expertise in his new role.”
Daniel Swisher, newly appointed Chief Executive Officer of NodThera, said: “NodThera is leading the NLRP3 inflammasome space with its best-in-class molecules and a wealth of pre-clinical and clinical data that is setting the tone for the field. NodThera has the potential to drive a positive and meaningful difference to the lives of patients suffering with obesity, cardiometabolic and neurodegenerative diseases and I am delighted to be leading the team at such an exciting point in the Company’s development, ahead of the readout from our ongoing cardiovascular risk trial in an inflamed obese population. I look forward to working alongside Alan, the Board and the wider team to achieve NodThera’s full potential.”
Alan Watt, President of Clinical & Preclinical R&D of NodThera, said: “I would like to extend the warmest welcome to Dan, who joins us at an exciting and pivotal moment. Our sector leading research in the field of NLRP3 inflammasome inhibition, our pioneering and demonstrated approach to reversal of neuroinflammation in humans and the strength of the NodThera team, provide an opportunity to bring about profound change in the management of obesity and multiple cardiometabolic and neurodegenerative diseases. I have the utmost confidence that, together, we will deliver on this opportunity.”
For more information about NodThera please contact:
NodThera
Tel: +44 (0) 1223 608130
Email: info@nodthera.com
ICR Consilium
Amber Fennell, David Daley, Sukaina Virji
Tel: +44 (0)20 3709 5700
Email: nodthera@consilium-comms.com
About NodThera
NodThera is a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases. Led by an experienced management team, NodThera is combining a deep understanding of NLRP3 inhibition, pharmaceutical neuroscience expertise and precision molecular chemistry. Its two lead clinical candidates are oral, small molecule NLRP3 inflammasome inhibitors, which have demonstrated differentiated, potentially best-in-class clinical profiles with significant anti-inflammatory effects and the ability to penetrate different areas of the brain, offering distinct opportunities to treat multiple indications. The Company is backed by top-tier investors including 5AM Ventures, Cowen Healthcare Investments, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Boston, MA, with additional operations in Cambridge, UK and Seattle, WA. Learn more at www.nodthera.com or follow the Company on LinkedIn.
- Sanofi receives exclusive rights to commercialize losmapimod in all territories outside the U.S.; Fulcrum retains full U.S. commercialization rights
- Fulcrum will receive an upfront payment of $80.0 million, and is eligible to receive $975.0 million in potential milestones, plus royalties on ex-U.S. product sales; parties will share future global development costs 50:50
- Conference call and webcast scheduled for 8:00 a.m. ET today to discuss the collaboration and other recent corporate developments, in conjunction with the first quarter 2024 financial results
CAMBRIDGE, Mass., May 13, 2024 (GLOBE NEWSWIRE) -- Fulcrum Therapeutics, Inc.® (Fulcrum) (Nasdaq: FULC), a clinical-stage biopharmaceutical company focused on developing small molecules to improve the lives of patients with genetically defined rare diseases, today announced that it has entered into a collaboration and license agreement with Sanofi (Nasdaq: SNY) for the development and commercialization of losmapimod, an oral small molecule being investigated for the treatment of facioscapulohumeral muscular dystrophy (FSHD). Under the collaboration and license agreement, Sanofi obtains exclusive commercialization rights for losmapimod outside of the U.S.
The collaboration and license agreement combines Fulcrum’s expertise in FSHD with Sanofi’s global reach and unparalleled commitment to treating patients with rare diseases. Losmapimod is currently being evaluated in a global Phase 3 clinical trial for the treatment of FSHD, a chronic and progressive genetic muscular disorder that is characterized by significant muscle cell death and fat infiltration into muscle tissue. Results from ReDUX4, the Phase 2 clinical trial evaluating losmapimod for the treatment of FSHD, demonstrated a slowing of disease progression and improved muscle health. Fulcrum expects to report topline data from REACH, the global Phase 3 clinical trial, in the fourth quarter of 2024. Following positive data from the Phase 3 trial, Fulcrum and Sanofi plan to submit marketing applications in the U.S., Europe, Japan, and other geographies.
“Sanofi is a proven leader in developing therapeutics for rare neuromuscular diseases and is the ideal partner to maximize the opportunity and reach of losmapimod outside the U.S.,” said Alex C. Sapir, Fulcrum’s president and chief executive officer. “This deal aligns with our core strategy, allowing Fulcrum to remain focused on preparations for commercialization of losmapimod in the U.S., while leveraging Sanofi’s exceptional global commercial capabilities and established infrastructure in key markets around the world. We are excited about the potential to provide the first approved treatment for FSHD patients, and we look forward to working with Sanofi to bring losmapimod to patients globally.”
“This partnership provides an exciting opportunity to expand Sanofi’s rare disease franchise and deliver the first approved FSHD treatment to patients with the strength and reach of our commercial organization,” said Burcu Eryilmaz, Sanofi’s Global Head of Rare Diseases. “Losmapimod has shown meaningful clinical benefits that underscore the disease-modifying potential and opportunity to address the high unmet need for a safe and effective drug that slows disease progression. With a deep commitment to bringing hope and new treatment options to patients, we look forward to working closely with Fulcrum as losmapimod advances through late-stage development.”
Per the terms of the agreement, Fulcrum will receive an upfront payment of $80.0 million and is eligible to receive up to an additional $975.0 million in specified regulatory and sales-based milestones, along with tiered escalating royalties starting in the low-teens on annual net sales of losmapimod outside the U.S. In addition, Fulcrum and Sanofi will equally share future global development costs.
Conference Call and Webcast
Individuals may register for the conference call by clicking the link here. Once registered, participants will receive dial-in details and unique PIN which will allow them to access the call. An audio webcast will be accessible through the Investor Relations section of Fulcrum’s website at www.fulcrumtx.com or by clicking here. Following the live webcast, an archived replay will also be available.
About Losmapimod
Losmapimod is a selective p38α/β mitogen activated protein kinase (MAPK) inhibitor. Fulcrum exclusively in-licensed losmapimod from GSK following Fulcrum’s discovery of the role of p38α/β inhibitors in the reduction of DUX4 expression and an extensive review of known compounds. Results reported from the Phase 2 ReDUX4 trial demonstrated a slowing of disease progression and improved function, including positive impacts on upper extremity strength and functional measures supporting losmapimod’s potential to be a transformative therapy for the treatment of FSHD. Although losmapimod had never previously been explored in muscular dystrophies, it had been evaluated in more than 3,600 subjects in clinical trials across multiple other indications, with no safety signals attributed to losmapimod. Losmapimod has been granted U.S. Food and Drug Administration (FDA) Fast Track designation and Orphan Drug Designation for the treatment of FSHD. Losmapimod is currently being evaluated in a Phase 3 multi-center randomized, double-blind, placebo-controlled, 48-week parallel-group study in people with FSHD (NCT05397470).
About FSHD
FSHD is a serious, rare, progressive and debilitating disease for which there are no approved treatments. It is characterized by fat infiltration of skeletal muscle leading to muscular atrophy involving primarily the face, scapula and shoulders, upper arms, and abdomen. Impact on patients includes profound decreases in the ability to perform activities of daily living, loss of upper limb function, loss of mobility and independence and chronic pain. FSHD is one of the most common forms of muscular dystrophy and has an estimated patient population of 30,000 in the United States alone.
About Fulcrum Therapeutics
Fulcrum Therapeutics is a clinical-stage biopharmaceutical company focused on developing small molecules to improve the lives of patients with genetically defined rare diseases in areas of high unmet medical need. Fulcrum’s two lead programs in clinical development are losmapimod, a small molecule in development for the treatment of facioscapulohumeral muscular dystrophy (FSHD), and pociredir (formerly known as FTX-6058), a small molecule designed to increase expression of fetal hemoglobin and in development for the treatment of sickle cell disease (SCD). Fulcrum uses proprietary technology to identify drug targets that can modulate gene expression to treat the known root cause of gene mis-expression. For more information, visit www.fulcrumtx.com and follow us on Twitter/X (@FulcrumTx) and LinkedIn.
Forward-Looking Statements
This press release contains “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995 that involve substantial risks and uncertainties. All statements, other than statements of historical facts, contained in this press release are forward-looking statements, including express or implied statements regarding Fulcrum’s collaboration and license agreement with Sanofi and receipt of the upfront payment thereunder; its ability to receive the milestone and royalty payments thereunder and achieve benefits therefrom; timing of data from REACH and its ability to support submission of marketing applications for losmapimod; and Fulcrum’s ability to deliver an FDA-approved therapy for FSHD patients; among others. The words “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “target,” “will,” “would” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. Any forward-looking statements are based on management’s current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in, or implied by, such forward-looking statements. These risks and uncertainties include, but are not limited to, risks associated with Fulcrum’s ability to continue to advance its product candidates in clinical trials; initiating and enrolling clinical trials on the timeline expected or at all; obtaining and maintaining necessary approvals from the FDA and other regulatory authorities; replicating in clinical trials positive results found in preclinical studies and/or earlier-stage clinical trials of losmapimod, pociredir and any other product candidates; obtaining, maintaining or protecting intellectual property rights related to its product candidates; managing expenses; managing executive and employee turnover, including integrating a new CMO; and raising the substantial additional capital needed to achieve its business objectives, among others. For a discussion of other risks and uncertainties, and other important factors, any of which could cause Fulcrum’s actual results to differ from those contained in the forward-looking statements, see the “Risk Factors” section, as well as discussions of potential risks, uncertainties, and other important factors, in Fulcrum’s most recent filings with the Securities and Exchange Commission. In addition, the forward-looking statements included in this press release represent Fulcrum’s views as of the date hereof and should not be relied upon as representing Fulcrum’s views as of any date subsequent to the date hereof. Fulcrum anticipates that subsequent events and developments will cause Fulcrum’s views to change. However, while Fulcrum may elect to update these forward-looking statements at some point in the future, Fulcrum specifically disclaims any obligation to do so.
Contact:
Chris Calabrese
LifeSci Advisors, LLC
ccalabrese@lifesciadvisors.com
917-680-5608
Inaugural list recognizes Cheung as one of the year’s most influential individuals in global health
Cambridge, MA – May 6, 2024 – NextPoint Therapeutics, a clinical-stage biotechnology company developing a new class of precision immuno-oncology and tumor-directed therapeutics targeting the novel B7-H7/HHLA2 axis, announced today that Chief Executive Officer Ivan Cheung has been named to the 2024 TIME100 Health List.
The inaugural TIME100 Health list recognizes the 100 most influential individuals in health and medicine who have made significant contributions to improving global health outcomes, advancing medical research and driving innovation in healthcare delivery.
“Ivan has demonstrated a relentless commitment to translating scientific breakthroughs into tangible solutions that benefit patients worldwide. The well-deserved recognition by TIME underscores his outstanding contributions to public health and exemplifies his visionary leadership and unwavering dedication to improving the lives of patients,” commented Detlev Biniszkiewicz, PhD, Managing Director at MPM BioImpact and Chairman of NextPoint’s Board of Directors. “Under Ivan’s leadership, NextPoint is advancing a novel class of therapeutics targeting the B7-H7 pathway to improve treatment options for cancer patients that do not respond to PD-1/L1 therapy.”
With over 25 years of experience leading drug development, approvals and launches, Mr. Cheung joined NextPoint as CEO in January 2024 aiming to deliver breakthrough clinical outcomes from a new world of precision immuno-oncology and tumor-targeting therapeutics. He previously served as Eisai’s U.S. CEO, where he guided the launch of the anti-cancer therapy lenvatinib in a range of tumor types. Importantly, Mr. Cheung led the first-ever full U.S. approval for lecanemab, a monoclonal antibody therapeutic to treat the underlying pathophysiology of early-stage Alzheimer’s disease and helped steward the reimbursement access from the Centers for Medicare & Medicaid Services for this class of medicines.
As a member of the TIME100 Health list, Mr. Cheung joins a distinguished group of individuals who are shaping the future of healthcare and driving positive change on a global scale. The full list and related tributes appear in the May 13, 2024 issue of TIME and at time.com/time100health.
About NextPoint Therapeutics
NextPoint is launching a new world of precision immuno-oncology and tumor-targeting therapeutics through its leading scientific work on the novel B7-H7/HHLA2 axis. Our innovative approach integrates foundational science with a defined clinical biomarker to identify the right patient population for each B7-H7-directed therapy, so that we can deliver a new class of monotherapies for patients including those who do not benefit from PD-1/L1 inhibitors. Our team of proven drug developers is simultaneously advancing therapeutic approaches blocking the B7-H7 immune signaling pathway and utilizing the unique upregulation of B7-H7 in cancer as an anchor for tumor-targeting treatment modalities. To learn more, visit nextpointtx.com.
Contacts
Media Contact
Chelsea Rule
MacDougall Advisors
1(781)235-3060

WILMINGTON, Del. & SAN DIEGO--(BUSINESS WIRE)--Apr. 23, 2024-- Incyte (Nasdaq:INCY) and Escient Pharmaceuticals, a clinical-stage drug discovery and development company advancing novel small molecule therapeutics for systemic immune and neuro-immune disorders, have entered into a definitive agreement under which Incyte has agreed to acquire Escient, including EP262, a first-in-class, potent, highly selective, once-daily small molecule antagonist of Mas-related G protein-coupled receptor X2 (MRGPRX2) and EP547, a first-in-class oral MRGPRX4 antagonist.
“As a company dedicated to innovation and the discovery of transformative medicines, we are excited to add EP262 and EP547 to our portfolio. This acquisition builds on our strategy to develop differentiated and first-in-class medicines with high potential,” said Hervé Hoppenot, Chief Executive Officer, Incyte. “EP262 and EP547 are complementary additions to our portfolio, providing an opportunity to leverage our expertise, address the needs of patients with inflammatory diseases and additional potential launch opportunities starting in 2029.”
By blocking MRGPRX2 and degranulation of mast cells, EP262 has the potential to effectively treat multiple mast cell-mediated diseases including atopic dermatitis (AD), chronic inducible urticaria (CIndU) and chronic spontaneous urticaria (CSU). Preclinical studies presented at the American Academy of Dermatology annual meeting in March 2023 showed that EP262 improved AD-like skin lesions and markers of type 2 inflammation. Additionally, in a Phase 1 study of 64 healthy volunteers, EP262 was safe and well tolerated at all doses tested, with no serious or severe adverse events, no adverse events leading to discontinuation and no clinically meaningful adverse changes in safety laboratory parameters, vital signs or ECG parameters. Treatment-emergent adverse events for EP262 were mild, with an incidence that was lower than placebo (33.3% vs. 62.5%) and did not increase with dose.
“These drug candidates are the result of the highly innovative research performed by Escient’s employees and scientific collaborators,” said Joshua A. Grass, President and Chief Executive Officer, Escient. “With its experienced development and commercial teams in Inflammation and Autoimmunity and portfolio of commercial and development stage products, Incyte is well positioned to translate this new science into valuable medicines for patients.”
Under the terms of the agreement, Incyte will acquire Escient and its assets for $750 million plus Escient’s net cash remaining at the close of the transaction, subject to customary adjustments. The acquisition is subject to clearance under the Hart-Scott-Rodino Act, among other customary conditions, and will become effective promptly following the satisfaction or waiver of these conditions which is currently anticipated to be by the third quarter of 2024.
Centerview Partners LLC and Goldman Sachs & Co. LLC advised Escient on the transaction and Fenwick & West LLP acted as legal counsel for Escient. Covington & Burling LLP acted as legal counsel for Incyte.
Incyte Conference Call and Webcast
Incyte will hold a conference call and webcast this morning at 8:00 a.m. ET. To access the conference call, please dial 877-407-3042 for domestic callers or +1 201-389-0864 for international callers. When prompted, provide the conference identification number: 13746287. If you are unable to participate, a replay of the conference call will be available for 90 days. The replay dial-in number for the United States is 877-660-6853 and the dial-in number for international callers is +1 201-612-7415. To access the replay, you will need the conference identification number: 13746287.
The conference call will also be webcast live and can be accessed at investor.incyte.com.
About EP262
EP262 is a potent, highly selective once-daily small molecule antagonist of MRGPRX2, a receptor expressed on mast cells that is activated by numerous ligands, including many peptides released from sensory neurons as well as other cell types. In response to MRGPRX2 activation, mast cells release histamine, tryptase, chymase, chemokines and cytokines, which can cause itchy hives, angioedema, type 2 inflammation (through engagement of the adaptive immune system) and chronic pruritus and pain. Preclinical data demonstrate that, by blocking activation of MRGPRX2, EP262 has the potential to effectively treat a broad range of mast cell-mediated conditions, with an initial focus on chronic urticarias and atopic dermatitis.
About EP547
EP547 is a potent, highly selective antagonist that blocks the activation of MRGPRX4 by various bile acids, bilirubin and urobilin. By virtue of this disease-specific mechanism of action, EP547 has the potential to be a highly targeted and efficacious treatment for cholestatic and uremic pruritus.
About Chronic Urticaria
Chronic urticaria, defined as urticaria persisting for more than 6 weeks, manifests with very itchy hives that may vary in size and can significantly impact a patient’s quality of life by interfering with sleep and daily activities. Some patients with chronic urticaria may also develop swelling deeper under the skin or in other tissues (angioedema). There are two main forms of chronic urticaria. In chronic spontaneous urticaria (CSU), hives occur spontaneously, without known triggers. In chronic inducible urticaria (CIndU), hives are induced by specific triggers, such as cold exposure (cold urticaria) or touch (symptomatic dermographism), among others.
About Incyte
A global biopharmaceutical company on a mission to Solve On., Incyte follows the science to find solutions for patients with unmet medical needs. Through the discovery, development and commercialization of proprietary therapeutics, Incyte has established a portfolio of first-in-class medicines for patients and a strong pipeline of products in Oncology and Inflammation & Autoimmunity. Headquartered in Wilmington, Delaware, Incyte has operations in North America, Europe and Asia.
For additional information on Incyte, please visit Incyte.com or follow us on social media: LinkedIn, X, Instagram, Facebook, YouTube.
About Escient Pharmaceuticals
Escient Pharmaceuticals is a clinical-stage company focused on developing novel therapeutics to address a broad range of neurosensory-inflammatory disorders. The company’s pipeline includes two first-in-class small molecule antagonists targeting MRGPRX2 for the treatment of various mast cell mediated disorders and MRGPRX4 for cholestatic pruritus. Based in San Diego, California, Escient is led by an experienced management and scientific team and funded by top-tier life science investors.
Incyte Forward-looking Statements
Except for the historical information set forth herein, the matters set forth in this press release, including statements regarding the opportunities presented by this transaction; whether and when EP262 or EP547 will be approved for use; whether and when Incyte will bring EP262 or EP547 to market; the potential of EP262 or EP547 to treat patients with atopic dermatitis (AD), chronic inducible urticaria (CIndU) and chronic urticaria (CSU) or for any other indication; and the potential for Incyte to broaden its ability to bring new medicines to patients, contain predictions, estimates and other forward-looking statements.
These forward-looking statements are based on the Company's current expectations and are subject to risks and uncertainties that may cause actual results to differ materially, including unanticipated developments in and risks related to: unanticipated delays; further research and development and the results of clinical trials possibly being unsuccessful or insufficient to meet applicable regulatory standards or warrant continued development; the ability to enroll sufficient numbers of subjects in clinical trials; the Company’s dependence on its relationships with its collaboration partners; the efficacy or safety of the Company’s products and the products of the Company’s collaboration partners; the acceptance of the Company’s products and the products of the Company’s collaboration partners in the marketplace; market competition; sales, marketing, manufacturing and distribution requirements; and other risks detailed from time to time in the Company’s reports filed with the Securities and Exchange Commission, including its annual report filed on Form 10-K for the year ended December 31, 2023. The Company disclaims any intent or obligation to update these forward-looking statements.
Incyte Contacts:
Media
media@incyte.com
Investors
ir@incyte.com
Escient Contact:
ir@escientpharma.com

- The Extension Funds Nucleai to Advance Its First-in-Class Spatial AI Biomarker in Active Clinical Enrollment
- Investment Expands Deployments of AI-Powered Spatial Biomarker Technology, Strengthening Companion Diagnostics for ADCs, Bi-specifics, and Immunotherapies
- M Ventures is the corporate strategic venture arm of Merck KGaA, Darmstadt, Germany
CHICAGO--(BUSINESS WIRE)--Nucleai, a spatial AI biomarker company that deciphers cellular conversations and maps cellular interactions within tissue samples to predict therapeutic outcomes, has secured a $14 million investment led by M Ventures, the corporate venture capital arm of Merck KGaA, Darmstadt, Germany, and supported by existing investors, bringing the total funding to $60 million. The investment enables Nucleai to further deploy its AI algorithms for the prospective enrollment of patients in clinical trials – a first in the field and a significant advancement in personalized solutions tailored to the distinct needs of patients.
The funding exemplifies Nucleai’s growing momentum in the life sciences industry, evidenced by collaborations with over 60 percent of the top 20 biopharma companies, venture backing from renowned investors like Section 32 and Sanofi Ventures, and now with M Ventures. The funding will accelerate the deployment of Nucleai’s spatial analysis technology that transforms a static biopsy slide into a dynamic AI-guided action plan, empowering pathologists with the intelligence to anticipate and navigate complex diseases, like cancer, with unmatched precision.
By integrating AI algorithms for prospective patient enrollment in clinical trials, Nucleai is spearheading a first-in-field approach to personalized medicine. This process is crucial for enhancing the accuracy of treatment targeting and activation within the body, particularly for advanced therapeutics such as antibody-drug conjugates (ADCs) and bi-specifics. Nucleai's platform tailors therapy strategies to the intricate cellular landscape of each patient, optimizing clinical trial participant selection. This personalized approach not only improves the likelihood of successful clinical outcomes but also significantly contributes to the speed and efficiency of bringing new therapies to market.
Leveraging AI and machine learning (ML), Nucleai analyzes pathology images and spatial data at the cellular and tissue levels. The proprietary technology extracts detailed patterns and features from medical images, offering profound insights into the tumor microenvironment, cellular morphology, and spatial relationships between different cell types. These capabilities advance drug development, refine biomarker discovery, and improve the precision of therapeutic targeting, ultimately leading to more effective and personalized treatment options for patients.
Avi Veidman, CEO and Co-Founder of Nucleai, said, “M Ventures' investment boosts our ability to scale and deploy our spatial AI technology for patient enrollment in clinical trials and supports our work in the rapidly emerging areas of immunotherapies, antibody-drug conjugates, and bi-specifics. Our vision is that every next-generation therapeutic is accompanied with an AI-enabled companion diagnostic, ensuring that each patient's treatment pathway is informed and efficacious. This funding positions us to scale spatial AI, not just to intercept but anticipate the complex behavior of diseases.”
Noga Yerushalmi, Investment Director at M Ventures, who is appointed to the board of Nucleai, stated, “Nucleai’s technology stands out by being the first spatial AI tool used by pathologists for clinical trial patient selection that is directly connected to a drug development program, setting them apart from other companies. Nucleai’s biology-driven mindset and their multimodal approach, which combines traditional pathology data with spatial biology data, allows for more accurate predictions of treatment responses, aligning perfectly with our commitment to optimise speed of new therapies to reach patients."
By moving beyond traditional pathology's analog analysis, Nucleai leverages AI to create dynamic, digital maps from biopsy samples, uncovering the complex interactions at play within tissue. This innovation marks a shift in disease diagnosis and treatment, offering an in-depth contextual view that underpins the development of novel therapeutics.
About Nucleai
Nucleai is the leading AI-powered spatial biomarker company, driving innovation at the intersection of technology and healthcare. Leveraging military intelligence-grade geospatial analytical methods, it intercepts, interprets, and analyzes complex cellular conversations and spatially oriented interactions within tissue samples, translating them into actionable insights. Nucleai’s platform empowers pathologists and researchers with an AI-powered data-rich action plan, paving the way for more informed decisions in the development of bi-specifics, antibody-drug conjugates (ADCs), and immunotherapy. Nucleai's investors include Section 32, Sanofi, Vertex Ventures, M Ventures, and Debiopharm Innovation Fund. It is headquartered in Israel and Chicago. For more information, please visit www.nucleai.ai.
About M Ventures
M Ventures is the strategic, corporate venture capital arm of Merck KGaA, Darmstadt, Germany. From its headquarters in the Netherlands and offices in Germany, USA and Israel, M Ventures invests globally in transformational ideas driven by innovative entrepreneurs. Taking an active role in its portfolio companies, M Ventures teams up with management teams and co-investors to translate scientific discoveries into commercial success. M Ventures focuses on identifying and financing novel solutions to some of the most difficult challenges, through company creation and equity investments in fields that will impact the vitality and sustainability of Merck KGaA, Darmstadt, Germany's current and future businesses.
Contacts
Consort Partners for Nucleai
nucleai@consortpartners.com
Today is a monumental day for Click, and I could not be more thrilled to share this news with you. Rejoyn™, originally developed as CT-152, is now THE FIRST prescription digital therapeutic to receive FDA clearance for the treatment of Major Depressive Disorder (MDD) symptoms. Rejoyn™ is indicated as a prescription digital therapeutic for the treatment of Major Depressive Disorder (MDD) symptoms as an adjunct to clinician-managed outpatient care for adult patients with MDD age 22 years and older who are on antidepressant medication. It is intended to reduce MDD symptoms. As first in its device type, Rejoyn™ was also assigned a new product code by FDA: “SAP - computerized behavioral therapy to treat major depressive disorder."
This FDA clearance represents an example of how digital therapeutics are positioned to help define the future of clinical care. The MIRAI study validated Rejoyn’s™ novel treatment approach and was central to the FDA filing. It is one of the largest studies completed to date on a digital therapeutic and one of the very few to evaluate a digital therapeutic’s effectiveness in a blinded comparison to a sham app that was designed to match the treatment in time, attention, and participant expectation of therapeutic effect.
The robust randomized controlled trial provided consistent evidence of effectiveness from both clinician and patient perspectives giving confidence in the benefit of Rejoyn™ as an adjunctive option for the treatment of MDD symptoms. The effectiveness was supported by a demonstrated safety profile with no treatment-emergent adverse events assessed as related to Rejoyn™ in the trial. Together the clinical trial data demonstrate that Rejoyn™ offers a novel adjunctive therapeutic option to treat MDD symptoms for adult patients who are experiencing an inadequate response to their antidepressant medication. And importantly, the Rejoyn™ data show that a prescription digital therapeutic can demonstrate a benefit beyond what patients were able to achieve with their existing antidepressant medication.
Quite simply, Rejoyn™ has the potential to help shift the course of mental health treatment by being the first to introduce the concept of a prescription digital therapeutic for the adjunctive treatment of MDD symptoms into the clinical mainstream at scale. We believe this product could mark a turning point toward a future in which clinically validated prescription software is a routine part of the standard of care.
I’m incredibly proud of the teams at Click, Otsuka and Mount Sinai who worked together to achieve today’s news, which is the culmination of years of effort. Congratulations to all involved!
This news comes at a very exciting time for our industry given the FDA Draft Guidance released last September on Prescription Drug Use-Related Software (PDURS) formally clarified that any added clinically meaningful benefit from the use of software together with a drug can be added directly to the drug label, right in the package insert. Soon, combination products developed using PDURS will unlock drug-like reimbursement via existing access pathways and extend the benefits of digital therapies to any therapeutic area with unmet behavioral, neurological, or cognitive needs where the drug alone may not be enough.
The future ahead is an exciting one, that if approached right, will enable a vibrant and evolving ecosystem of digital treatment options for patients. We are passionate about this vision and could not be more excited to make this future a reality. We are grateful for our collaborators, dedicated investors, and many exceedingly talented colleagues who have made today’s news possible. And if you haven’t already, we invite you to please join us – the opportunity to improve patient care with digital therapeutics is just beginning.
You can read more about the clinical data for Rejoyn™ in the Clinician Brief Summary; a link is provided below.
https://rejoynhcp.com/Clinician-Brief-Summary.pdf
You can read more about the FDA Clearance in the press release; a link is provided below.
https://www.otsuka-us.com/news/rejoyn-fda-authorized
With warm regards,
David Benshoof Klein
CEO, Click Therapeutics
INDICATION and SAFETY INFORMATION for Rejoyn™
INDICATION:
Rejoyn™ is a prescription digital therapeutic for the treatment of Major Depressive Disorder (MDD) symptoms as an adjunct to clinician-managed outpatient care for adult patients with MDD age 22 years and older who are on antidepressant medication. It is intended to reduce MDD symptoms.
SAFETY INFORMATION:
Rejoyn™ is not intended to be used as a standalone therapy or a substitute for medication. Patients should continue their current treatment as directed. Rejoyn™ does not monitor the patient’s symptoms or clinical status and cannot send or receive alerts or warnings to the prescriber.
Patients should be clearly instructed that if they believe their depression is worsening or if they have feelings or thoughts of harming themselves or others, to contact a healthcare professional, dial 911, or go to the nearest emergency room immediately.

- Rapidly advancing multiple therapeutic programs leveraging Mirador360 TM precision development engine incorporating advances in human genetics and cutting-edge data science
- Company led by several executives of Prometheus Biosciences and founded by chairman and CEO Mark C. McKenna
- Over $400 million in financing led by ARCH Venture Partners with early investments from OrbiMed and Fairmount and participation from other premier life sciences investors
SAN DIEGO, March 20, 2024 -- Mirador Therapeutics, Inc. (Mirador), announced its launch today. Founded by Mark C. McKenna and led by several former executives of Prometheus Biosciences (acquired in 2023 by Merck for $10.8 billion), Mirador aims to revolutionize precision medicine for immune-mediated inflammatory and fibrotic diseases by leveraging its proprietary Mirador360TM development engine to rapidly advance multiple programs. Mirador has raised more than $400 million in financing led by ARCH Venture Partners, with early investments from OrbiMed and Fairmount. Other premier life sciences investors also participated, including Fidelity Management & Research Company, Point72, Farallon Capital Management, Boxer Capital, TCGX, Invus, Logos Capital, Moore Strategic Ventures, Blue Owl Healthcare Opportunities, Sanofi Ventures, Woodline Partners LP, Venrock Healthcare Capital Partners, RTW Investments and Alexandria Venture Investments.
“At Mirador, we envision a bold new era of precision medicine for immune-mediated inflammatory and fibrotic diseases driven by speed and superior development accuracy,” said Mark C. McKenna, chairman and CEO of Mirador. “The industry has only scratched the surface of utilizing advances in human genetics – coupled with exponential progress in machine learning – to accelerate the development of precision therapies for patients who need them the most. With a proven team, distinguished board of directors, leading healthcare investors and proprietary data-driven approach, we aim to create a leading precision medicine company at scale to provide important new treatment options for patients.”
“The I&I field is in need of better, novel therapeutics as well as new R&D approaches that target enriched patient populations for improved probability of success in the clinic,” said Kristina Burow, Mirador board member and managing director of ARCH Venture Partners. “The Mirador team has an outstanding track record of success in precision immunology, and we are well on our way to building a company that will make a lasting impact on the lives of millions of patients suffering from a broad range of immune-mediated inflammatory and fibrotic diseases.”
The Next Wave of Precision Medicine in Immunology and Inflammation (I&I)
Mirador’s focus is on developing first-in-class or best-in-class precision medicines. To accelerate development, Mirador utilizes Mirador360, its proprietary precision development engine that leverages recent breakthroughs in human genetics and cutting-edge data science. Mirador360 is purpose-built to harmonize millions of patient samples to discover and validate genetic associations to immuno-fibrotic diseases, identify novel therapeutic targets and elucidate target-target interactions as well as optimal target-target pairs for potential combination therapies. Mirador360 also allows Mirador to develop diagnostics and stratify heterogeneous patient populations for precise clinical development.
About Mirador Therapeutics
Mirador is a next-generation precision medicine company focused on immunology and inflammation. The company’s Mirador360 TM precision development engine leverages the latest advances in human genetics and cutting-edge data science to rapidly advance new precision medicines for patients living with chronic immune-mediated inflammatory and fibrotic diseases. Launched in 2024, Mirador has raised over $400 million from leading life sciences investors and is based in San Diego, CA. For more information, please visit us at www.miradortx.com and follow us on Linkedin.
Contacts:
Media:
Dan Budwick
1AB
dan@1abmedia.com
Investors:
Steve Klass
1AB
steve@1abmedia.com
- Brain-penetrant NLRP3 inflammasome inhibitor NT-0796 reduced key neuroinflammatory and inflammatory biomarkers in Parkinson’s disease patients to the levels found in healthy elderly controls over 28 days
- Demonstrates potential to change the treatment paradigm and halt disease progression with a disease-modifying approach
- Trial results will be presented at AD/PDTM 2024 on Friday March 8 in Lisbon, Portugal
- Preparations for a Phase IIa/IIb study in Parkinson’s disease are ongoing
BOSTON, MA, March 7, 2024 - NodThera, a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases, today announces positive data from its Phase Ib/IIa study in Parkinson’s disease patients, evaluating the effects of its oral, brain-penetrant NLRP3 inflammasome inhibitor NT-0796, on inflammatory and disease-specific biomarkers in the blood and cerebrospinal fluid (CSF).
NodThera’s study demonstrated mean reductions of key pro-inflammatory biomarkers in CSF (e.g. IL-1β, IL-6, CCL2, CXCL1 and CXCL8) over 28 days compared to baseline to levels approximating those of healthy elderly controls, demonstrating reversal of NLRP3-mediated neuroinflammation. In addition, reductions in neurodegenerative markers were also observed following oral dosing of NT-0796, including NfL and soluble TREM (sTREM2). In subjects with elevated acute phase biomarkers CRP and fibrinogen, levels were reduced significantly, consistent with the peripheral anti-inflammatory effects of NT-0796 seen in elderly healthy volunteers studied in an earlier stage of the trial.
Alan Watt, Chief Executive Officer of NodThera, said: “Our new findings in Parkinson’s disease, a condition with substantial unmet medical needs, are profound and highly encouraging. They bolster our confidence in the ongoing program, with Phase II studies now in advanced planning stages. The correlation between Parkinson’s disease and neuroinflammation is well-documented, with alpha-synuclein fibrils triggering microglial NLRP3 activation, leading to neuroinflammation and subsequent neurodegeneration. This is the inaugural demonstration of an NLRP3 inhibitor’s potential to not only address Parkinson’s disease but also offer a broader impact on neurodegenerative diseases. Given that existing Parkinson’s treatments primarily manage symptoms, our innovative, disease-modifying strategy presents a significant shift, aiming to stop the disease progression. NT-0796’s demonstrated efficacy in reducing neuroinflammation in patients heralds a substantial advancement towards halting this devastating disease.”
NT-0796 was safe and well tolerated; adverse events (AEs) were mainly mild and transient, and no serious adverse events (SAEs) were observed. Pharmacokinetics indicated the drug candidate’s potential for once-daily dosing and levels of the drug candidate in the brain, determined by measuring the concentration in CSF, were found to be maintained over 24 hours.
Taken together, the findings demonstrate that NT-0796 successfully delivered anti-neuroinflammatory and anti-inflammatory changes within 7 days and sustained for 28 days, indicating excellent potential for long-term, oral dosing in Parkinson’s disease patients.
Prof. Thomas Foltynie BSc, MBBS, MRCP, PhD, Professor of Neurology in the Department of Clinical and Movement Neurosciences, UCL Institute of Neurology and Consultant Neurologist at the National Hospital for Neurology and Neurosurgery, Queen Square, London, said: “These results are particularly promising for the future of Parkinson’s disease treatment, providing a compelling approach to the modulation of inflammation in the brain, a key driver in the development of this disease. Such an approach has the potential to change the face of treatment – actually stopping the disease in its tracks, that would be of enormous value to those living with Parkinson’s disease.”
The findings from NodThera’s study will be presented on Friday March 8 at AD/PDTM 2024, the international conference on Alzheimer’s and Parkinson’s diseases and related neurological disorders, taking place in Lisbon, Portugal.
Additionally, following the recently published preclinical data demonstrating NodThera’s NLRP3 inflammasome inhibitors reversed diet-induced obesity (DIO) and inflammation in mice the Company’s pioneering Phase Ib/IIa clinical study of NT-0796 in obese subjects with cardiovascular risk is in progress with results expected by end of 2Q24. This biomarker-rich study is measuring the change in baseline to Day 28 of CRP levels, a key peripheral inflammatory marker and known predictor of risk of developing atherosclerotic cardiovascular (CV) disease as well as the potential for modification of body weight over 28 days.
For more information about NodThera please contact:
NodThera
Tel: +44 (0) 1223 608130
Email: info@nodthera.com
ICR Consilium
Amber Fennell, David Daley, Sukaina Virji
Tel: +44 (0)20 3709 5700
Email: nodthera@consilium-comms.com
About NodThera
NodThera is a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases. Led by an experienced management team, NodThera is combining a deep understanding of NLRP3 inhibition, pharmaceutical neuroscience expertise and precision molecular chemistry. Its two lead clinical candidates are oral, small molecule NLRP3 inflammasome inhibitors, which have demonstrated differentiated, potentially best-in-class clinical profiles with significant anti-inflammatory effects and the ability to penetrate different areas of the brain, offering distinct opportunities to treat multiple indications. The Company is backed by top-tier investors including 5AM Ventures, Blue Owl Capital, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Boston, MA, with additional operations in Cambridge, UK and Seattle, WA. Learn more at www.nodthera.com or follow the Company on LinkedIn.
UNC13A is an essential regulator of neurotransmitter release at synapses; mis-splicing is a critical RNA alteration occurring in up to 63 percent of all ALS patients and up to one-third of all FTD cases
Preclinical data to be featured in a poster presentation at AD/PD™ 2024
CAMBRIDGE, Mass., March 4, 2024 – QurAlis Corporation, a clinical-stage biotechnology company driving scientific breakthroughs into powerful precision medicines that will alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative diseases, today announced preclinical data that showed the company’s UNC13A splice-switching antisense oligonucleotides (ASOs) modulate UNC13A splicing and restore normal synaptic activities in ALS and FTD. QurAlis’ ASOs prevented cryptic exon inclusion in UNC13A transcripts, increased UNC13A protein levels, and normalized localization of UNC13A protein at the synapse.
Amyotrophic lateral sclerosis is a progressive neurodegenerative disease characterized by the loss of neurons in the spinal cord, brainstem, and brain. A defining feature of both sporadic and familial disease is the cytoplasmic mis-localization of TAR DNA Binding Protein-43 (TDP-43). TDP- 43 pathology is implicated in 95 percent of ALS cases and 50 percent of FTD cases.
UNC13A is an essential regulator of neurotransmitter release at synapses and is one of a number of pre-mRNAs that becomes mis-spliced due to loss of nuclear TDP-43 in disease. Up to 63 percent of ALS patients and up to one-third of FTD patients carry a single nucleotide polymorphism in the UNC13A gene or show TDP-43 pathology which greatly exacerbates UNC13A mis-splicing leading to loss of function of the UNC13A protein.
“There are currently no cures for ALS or FTD and limited therapeutic options are available for ALS and FTD patients who are in desperate need for effective therapies,” said Dan Elbaum, Ph.D., chief scientific officer of QurAlis. “QurAlis has identified ASOs that modulate UNC13A splicing and restore normal synaptic activities. We believe that correction of UNC13A splicing can provide therapeutic benefit in relevant patient populations.”
These data will be featured in a poster presentation at the AD/PD™ 2024 International Conference on Alzheimer’s and Parkinson’s Diseases and Related Neurological Disorders being held March 5-9, 2024 in Lisbon, Portugal. Details of the presentation are as follows:
Title: UNC13A Targeting Splice Switching ASOs Ameliorate TDP-43 Dependent Mis-splicing Phenotypes in FTD and ALS
Date(s)/Time: Thursday, March 7, 2024, and Friday, March 8, 2024 at 1:50-3:50PM CET
Poster and Abstract Number: P1214 / 2851
Poster Topic: FTD, ALS: TDP43, C9orf72, and TMEM106B 1 Session: D02 Therapeutic Targets, Mechanisms for Treatment Location: Auditorium VIII
Presenter: Marisa Kamelgarn, Ph.D., senior scientist, QurAlis
Splice-switching oligonucleotides targeting UNC13A were screened via quantitative polymerase chain reaction (qPCR) and a small subset demonstrated correction of splicing and rescue of protein. From those rescue experiments, ASOs were used for functional assay testing.
Using in-house models of induced pluripotent stem cell (iPSC) motor and cortical neurons, QurAlis established a phenotype to demonstrate the cellular consequences of UNC13A mis- splicing due to TDP-43 loss of function.
TDP-43 mediated UNC13A loss partially disabled SNARE complex assembly and evoked synaptic vesicle release in human motor neurons. QurAlis’ ASOs restored all or some aspects of SNARE complex assembly and synaptic vesicle fusion and release.
About QurAlis’ FlexASO™ Splice Modulator Platform
Incorporating its proprietary FlexASO™ Splice Modulator Platform, QurAlis’ ASOs correct UNC13A mis-splicing, restore UNC13A protein production, and reduce cryptic exons that may contribute to disease progression. The QurAlis FlexASO Splice Modulator Platform was developed to generate splice-switching ASOs with improved potency and an increased therapeutic index. In addition to UNC13A, QurAlis is currently exploring this ASO technology for multiple other disease targets.
About QurAlis Corporation
At QurAlis, we are neuro pioneers on a quest to cure. We work with a relentless pursuit of knowledge, a precise attention to craft, and an optimistic mindset to discover and develop effective precision medicines that will alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative diseases. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a pipeline with therapeutic candidates that target specific components of ALS and FTD pathology and defined patient populations based on both disease-causing genetic mutation(s) and clinical biomarkers. For more information, please visit www.quralis.com or follow us on Twitter @QurAlisCo.
Media contact:
Kathy Vincent
kathy@kathyv ncent.com
310-403-8951
EpiMonitor is an all-in-one epilepsy monitoring solution that uses the EmbracePlus wearable and a mobile app to detect and alert for seizures, while providing patients and physicians with continuous, accurate health insights
BOSTON--(BUSINESS WIRE)--Empatica, a leader in remote health monitoring powered by AI, is proud to announce the official launch of EpiMonitor, the latest advancement in wearable epilepsy monitoring.
EpiMonitor has been FDA-cleared for use in adult and children populations aged 6 and up, and is the only wearable solution with regulatory approval for seizure detection available to purchase in the US. It is the successor to Embrace2, which enjoyed global acclaim as the leading wearable for seizure detection for years, and the first ever to be available to patients.
EpiMonitor represents a significant leap forward in epilepsy monitoring technologies, thanks to enhanced capabilities including new seizure alert features, up to a week of battery life, and advanced health insights. It utilizes Empatica’s next-generation medical watch, EmbracePlus, which has already been successfully used to monitor the health of thousands of participants in hundreds of clinical trials. Together with a companion smartphone app and smart algorithms, EpiMonitor is a complete system that continuously monitors its wearer’s health data, detecting possible generalized tonic-clonic seizures, alerting caregivers, and providing valuable health information to help users better manage their condition.
"We are thrilled to introduce EpiMonitor to people with epilepsy in the US,” said Empatica’s Chief Scientist and Co-founder, Dr. Rosalind Picard. “Empatica has always provided the only smartwatch seizure monitors validated by the FDA. EpiMonitor takes this to a new level: giving patients up to 7 days of battery life, and new features that give them greater control over seizure alerts. Also, at the touch of a button, patients can now see comprehensive health insights – including seizures, sleep, and activity reports – in a form easy to give their clinician. EpiMonitor is the device we've long dreamed of to open a new era in giving epilepsy patients greater control over their lives.”
Key features of EpiMonitor include:
- FDA-cleared for adults and children ages 6 and up
- Automatic seizure detection through a smart algorithm (98% accuracy; low False Alarm Rate) and alerts to caregivers via a call and SMS with the user’s location
- Up to 7 days of battery life
- Advanced sensor technology for accurate data collection
- Option to send self-triggered alerts to caregivers
- A user-friendly mobile app with a seizure diary
- Sleep and activity tracking through Empatica’s algorithms
- Seizure, sleep and activity reports that can be exported and shared with a physician
- iOS and Android compatibility
The EpiMonitor FDA clearance also represents a significant milestone for Empatica as it demonstrates the successful integration of a new algorithm with the existing technology stack of the Empatica Health Monitoring Platform, highlighting its potential to host future SaMD products.
Seizure forecasting
In December 2023, Empatica also announced plans to conduct a groundbreaking study to develop a seizure forecasting algorithm, based on the world’s largest real-world data set in epilepsy patients. Recruitment for the study is available to all US-based EpiMonitor users.
“Our FORESIGHT clinical study is an incredibly exciting opportunity to advance personalized seizure forecasting,” said Dr. Marisa Cruz, Empatica’s Chief Medical Officer. “This research is a critical step in our ongoing development of innovative and effective seizure management tools that improve clinical outcomes and quality of life for patients with epilepsy.”
By enrolling in the study, users can contribute valuable data to ongoing research efforts, and participate in a large-scale scientific effort to create a better tomorrow for people living with epilepsy. Large datasets are extremely important to train an algorithm to achieve a prediction of when someone will have a seizure, but they are also extremely difficult to gather. With this study, Empatica seeks to overcome this challenge with the participation of its community.
Seizure forecasting can have multiple benefits, including early intervention in the form of tailoring therapies to the start of seizure onset1, better seizure management and treatment2, and useful information that can be used to modify daily activities2,3.
EpiMonitor is available for purchase in the US from the Empatica website. To learn more visit empatica.com/epimonitor.
Empatica Inc is a pioneer in continuous, unobtrusive remote health monitoring driven by AI. Empatica's platform and technology are used by thousands of institutional partners for research purposes, in studies examining stress, sleep, epilepsy, migraine, depression, addiction, and other conditions. In 2018, Empatica’s Embrace wearable received FDA clearance for seizure monitoring, making it the world's first epilepsy watch to be cleared by the FDA. Its latest flagship medical wearable, EmbracePlus, has been developed with key partners including HHS, USAMRDC, and NASA.
1 Epilepsy Foundation (https://www.epilepsy.com/research-funding/epilepsy-innovation-institute/seizure-gauge-challenge)
2 https://www.nature.com/articles/s41598-021-01449-2
3 https://pubmed.ncbi.nlm.nih.gov/33040327/
Contacts
Marianna Xenophontos
mx@empatica.com
- Data published in Journal of Pharmacology and Experimental Therapeutics
- NodThera’s brain-penetrant NLRP3 inhibitors matched weight loss driven by GLP-1 receptor agonist semaglutide (Wegovy®) or calorie restriction, while also providing enhanced improvements in disease-relevant biomarkers of cardiovascular inflammation and lipid metabolism
- Findings indicate NLRP3 activation in the brain is implicated in driving obesity which can be reversed with brain-penetrant NLRP3 inhibitors
- Additionally, combinations of an NLRP3 inhibitor and a GLP-1 receptor agonist were shown to be additive on weight loss in as-yet unpublished data
- NodThera’s lead candidate NT-0796 is currently in Phase Ib/IIa development in cardiometabolic disease and Parkinson’s disease
BOSTON, MA, February 19, 2024 – NodThera, a leading clinical-stage biotech developing brain-penetrant NLRP3 inhibitors to treat chronic inflammatory diseases, today announces the publication of preclinical data demonstrating its clinical-stage investigational compounds reversed diet-induced obesity (DIO) and inflammation in an animal model of disease.
The data are published in the Journal of Pharmacology and Experimental Therapeutics in a paper titled ‘Reversal of high fat diet-induced obesity, systemic inflammation and astrogliosis by the NLRP3 inflammasome inhibitors NT-0249 and NT-0796’1.
The NLRP3 inflammasome is a highly validated anti-inflammatory drug target, and these findings demonstrate that NLRP3 plays a key role in controlling obesity and obesity-associated inflammation through the modulation of hypothalamic gliosis. Both NT-0796 and NT-0249, two structurally distinct NLRP3 inhibitors in clinical development by NodThera, have generated a wealth of preclinical and clinical data demonstrating brain-penetration and broad anti-inflammatory effects, with NT-0796 being the first NLRP3 inhibitor to show reduced neuroinflammation in the clinic.
In this latest publication, NodThera’s researchers show for the first time the ability of NT-0796 and NT-0249 to reverse DIO in a murine model, providing comparisons against the effects of the GLP-1 receptor agonist (GLP-1RA) semaglutide (Wegovy®) and calorie restriction. While all three therapeutic approaches led to statistically significant reductions in body fat in DIO mice, only the NLRP3 inhibitors reduced disease-relevant cardiovascular inflammatory biomarkers such as fibrinogen, sVCAM-1, suPAR, and PCSK9, suggesting their potential to further reduce cardiovascular risk in obese populations. NodThera has additionally explored alternative treatment scenarios where NLRP3 inhibitors can be combined with a GLP1-RA or used as a follow-on therapy for patients who do not tolerate GLP-1RA drugs. Yet-to-be-published preclinical findings have demonstrated an additive weight loss effect when combining the brain-penetrant NLRP3 inhibitors with low dose GLP-1RAs, and stable weight maintenance following cessation of GLP-1RA therapy by dosing of a brain-penetrant NLPR3 inhibitor, thereby preventing body mass regain.
Obesity is a global health concern that predisposes individuals to chronic disease such as diabetes and cardiovascular disease at least in part by promoting systemic inflammation.
Alan Watt, Chief Executive Officer of NodThera, said: “These remarkable findings – the first in the field of NLRP3 inflammasome research – suggest that in obese mice consuming a high-fat diet, brain- penetrant NLRP3 inhibition and the resulting anti-inflammatory effect confers not only reversal of obesity but metabolic benefits that extend well beyond this. While GLP-1 receptor agonists have undoubtedly delivered significant achievements in the management of obesity, their adverse event profile is well established, presenting difficulties for some patients. Our brain-penetrant NLRP3 inhibitors deliver robust weight loss and broad cardiometabolic benefits by targeting a novel molecular mechanism with the convenience of oral dosing and an exceptional safety profile. Our ongoing Phase IIa study in obese individuals at cardiovascular risk will further validate these pre- clinical findings.”
References:
1. Reversal of High Fat Diet-Induced Obesity, Systemic Inflammation, and Astrogliosis by the NLRP3 Inflammasome Inhibitors NT-0249 and NT-0796. Peter Thornton, Valérie Reader, Zsofia Digby, Pamela Smolak, Nicola Lindsay, David Harrison, Nick Clarke and Alan P. Watt. Journal of Pharmacology and Experimental Therapeutics March 2024, 388 (3) 813-826; DOI: https://doi.org/10.1124/jpet.123.002013
For more information about NodThera please contact:
NodThera
Tel: +44 (0) 1223 608130
Email: info@nodthera.com
ICR Consilium
Amber Fennell, David Daley, Sukaina Virji
Tel: +44 (0)20 3709 5700
Email: nodthera@consilium-comms.com
About NodThera
NodThera is a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases. Led by an experienced management team, NodThera is combining a deep understanding of NLRP3 inhibition, pharmaceutical neuroscience expertise and precision molecular chemistry. Its two lead clinical candidates are oral, small molecule NLRP3 inflammasome inhibitors, which have demonstrated differentiated, potentially best-in-class clinical profiles with significant anti-inflammatory effects and the ability to penetrate different areas of the brain, offering distinct opportunities to treat multiple indications. The Company is backed by top-tier investors including 5AM Ventures, Blue Owl Capital, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Boston, MA, with additional operations in Cambridge, UK and Seattle, WA. Learn more at www.nodthera.com or follow the Company on LinkedIn.

Collaboration combines Intellia’s leading CRISPR-based platform, including its DNA writing technology, with ReCode’s proprietary Selective Organ Targeting (SORT) lipid nanoparticle (LNP) to extend the reach of gene editing to disease-causing targets in the lung
CAMBRIDGE, Mass. and MENLO PARK, Calif., Feb. 15, 2024 (GLOBE NEWSWIRE) -- Intellia Therapeutics, Inc. (NASDAQ:NTLA), a leading clinical-stage gene editing company focused on revolutionizing medicine with CRISPR-based therapies, and ReCode Therapeutics, a clinical-stage genetic medicines company using tissue-specific delivery to power the next wave of mRNA and gene correction therapeutics, today announced a strategic collaboration to develop novel genomic medicines for the treatment of cystic fibrosis (CF). CF is a genetic disease caused by mutations in the CFTR gene, leading to the accumulation of thick mucus in the lungs, digestive systems and other organs. CF can result in life-threatening infections, respiratory failure and other serious complications.
The collaboration will leverage Intellia’s proprietary CRISPR-based gene editing platform, including its DNA writing technology, and ReCode’s proprietary Selective Organ Targeting (SORT) lipid nanoparticle (LNP) delivery platform to precisely correct one or more CF disease-causing gene mutations. As part of the agreement, the companies will focus initial research efforts on therapeutic approaches that address CF for patients who have limited or no treatment options available, with the opportunity to expand the scope of the collaboration in later phases. Intellia will be responsible for the design of the editing strategy and research- grade components for the investigational therapies. ReCode will lead the subsequent preclinical and clinical development. ReCode will also lead worldwide commercialization for certain programs arising from the collaboration. Intellia will be eligible to receive pre-specified development and commercial milestone payments, as well as royalties on potential sales. Intellia may also exercise an option to lead commercialization in the U.S. for certain programs.
“Intellia’s vision to realize the full promise of gene editing includes extending the reach of our industry-leading CRISPR-based platform to targets outside the liver. This collaboration with ReCode is aimed at achieving that goal as we work together to accelerate the development of potentially life-changing therapies for people with cystic fibrosis,” said Intellia President and Chief Executive Officer John Leonard, M.D. “Building on our CRISPR/Cas9 capabilities, we have made important progress advancing our proprietary DNA writing technology to enable a range of precise editing strategies. We are excited to combine our gene editing expertise and platform with ReCode’s novel lung-directed LNP delivery platform.”
"We are excited to partner with Intellia, a clear leader in the gene editing space, with the ultimate goal of bringing life-altering therapies to CF patients,” said ReCode Chief Executive Officer Shehnaaz Suliman, M.D. (MB ChB), M.B.A., M.Phil. “This collaboration provides further validation of ReCode's SORT LNP platform to deliver diverse gene editing modalities to specific cells and tissues. By combining our highly synergistic technologies and capabilities, we are excited about the potential to enable a faster path for next-generation gene editing therapeutics to CF patients.”
About Intellia Therapeutics
Intellia Therapeutics, Inc. (NASDAQ:NTLA) is a leading clinical-stage gene editing company focused on revolutionizing medicine with CRISPR-based therapies. The company’s in vivo programs use CRISPR to enable precise editing of disease-causing genes directly inside the human body. Intellia’s ex vivo programs use CRISPR to engineer human cells outside the body for the treatment of cancer and autoimmune diseases.
Intellia’s deep scientific, technical and clinical development experience, along with its people, is helping set the standard for a new class of medicine. To harness the full potential of gene editing, Intellia continues to expand the capabilities of its CRISPR-based platform with novel editing and delivery technologies. Learn more at intelliatx.com and follow us @intelliatx.
About ReCode Therapeutics
ReCode Therapeutics is a clinical-stage genetic medicines company using precision delivery to power the next wave of mRNA and gene correction therapeutics. ReCode’s proprietary Selective Organ Targeting (SORT) lipid nanoparticle (LNP) platform enables highly precise and targeted delivery of genetic medicines directly to the organs, tissues and cells implicated in disease, enabling improved efficacy and potency. ReCode’s lead programs include RCT1100 for the treatment of primary ciliary dyskinesia caused by pathogenic mutations in the DNAI1 gene, and RCT2100 for the treatment of the 10-13% of cystic fibrosis patients who have Class I mutations in the CFTR gene and do not respond to currently approved CFTR modulators. RCT1100 and RCT2100 are inhaled disease-modifying mRNA-based therapies formulated using the SORT LNP delivery platform. ReCode is expanding its pipeline to develop potential therapies for other rare and common genetic diseases, including musculoskeletal, central nervous system, liver and infectious disease indications.
ReCode has been recognized by the San Francisco Business Times and Silicon Valley Business Journal as a Best Place to Work. For more information, visit www.recodetx.com and follow us on LinkedIn.
Intellia Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995, as amended, including, without limitation, express or implied statements regarding Intellia’s beliefs and expectations regarding: its strategy, business plans and focus; its ability to leverage its proprietary CRISPR-based gene editing platform, including its DNA writing technology, and to combine its platform with the proprietary lipid nanoparticle (“LNP”) delivery platform developed by ReCode Therapeutics, Inc. (“ReCode”) to precisely correct one or more cystic fibrosis (“CF”) disease-causing gene mutations; its ability to develop therapeutic approaches that address CF for patients who have limited or no treatment options available, and to expand the scope of the collaboration in later phases; its ability to commercialize in the U.S. the products that may result from its collaboration with ReCode; and the expected strategic benefits of any current or future collaborations, including its collaboration with ReCode.
Any forward-looking statements in this press release are based on management's current expectations and beliefs of future events, and are subject to a number of risks, uncertainties and important factors that may cause actual events or results to differ materially from those expressed or implied by any forward-looking statements contained in this press release, including, without limitation, risks related to Intellia’s ability to protect and maintain its intellectual property portfolio; risks related to Intellia’s relationship with third parties, including its licensors and licensees; risks related to the ability of Intellia’s licensors to protect and maintain their intellectual property position; uncertainties related to the development of the company’s product candidates, including product candidates to be developed in its collaboration with ReCode, and the authorization, initiation and conduct of studies and other development requirements for such product candidates; the risk that any one or more of Intellia’s or its collaborators’ product candidates (including the product candidates to be developed with ReCode) will not be successfully developed and commercialized; the risk that the results of preclinical studies or clinical studies will not be predictive of future results in connection with future studies; and the risk that Intellia’s collaboration with ReCode or its other collaborations will not continue or will not be successful. These and other risks and uncertainties are described in greater detail in Intellia’s other filings with the Securities and Exchange Commission, including the section entitled “Risk Factors” in Intellia’s most recent annual report on Form 10-K and quarterly report on Form 10-Q, as well as discussions of potential risks, uncertainties, and other important factors in other filings. Any forward-looking statements contained in this press release represent Intellia’s views only as of the date hereof and should not be relied upon as representing its views as of any subsequent date. Intellia explicitly disclaims any obligation to update any forward-looking statements, except as required by law.
Intellia Contacts:
Investors:
Ian Karp
Senior Vice President, Investor Relations and Corporate Communications
ian.karp@intelliatx.com
Lina Li
Senior Director, Investor Relations and Corporate Communications
lina.li@intelliatx.com
Media:
Matt Crenson
Ten Bridge Communications
media@intelliatx.com
BCIntellia@tenbridgecommunications.com
ReCode Contacts:
Investors:
Anne Marie
Fields Stern IR
AnneMarie.Fields@sternir.com
IR@recodetx.com
Media:
Erica Jefferson
Senior Vice President, Corporate Affairs
ejefferson@recodetx.com
Tara Cooper
The Grace Group
tara@gracegroup.us
Cambridge, MA – February 14, 2024 – NextPoint Therapeutics, a clinical-stage biotechnology company developing a new class of precision oncology therapeutics targeting the novel HHLA2 pathway, announced today the closing of a $42.5M Extension to its Series B financing round resulting in a total of $122.5M raised in the Series B financing. The funds will be used to advance the company’s two immuno- oncology clinical programs, NPX267 and NPX887, as well as propel the development of additional therapeutic modalities in the pipeline that target the novel HHLA2 tumor antigen.
The Extension was led by existing investor Catalio Capital Management, and as part of the financing, R. Jacob Vogelstein, PhD, has joined the NextPoint Board of Directors. Catalio is joined by other existing investors including MPM BioImpact, Leaps by Bayer, Sanofi Ventures, Invus, Sixty Degree Capital, Dana- Farber Cancer Institute’s Binney Street Capital and NextPoint founder Gordon Freeman, PhD. Along with Catalio, new investors Arkin Bio-Capital and WTT investment Ltd were the largest participants in this fundraising.
“Catalio is committed to investing in the next generation of category-defining life sciences companies and NextPoint’s mission of delivering groundbreaking new options to more patients with cancer strongly resonates with our own,” said R. Jacob Vogelstein, PhD, Co-Founder & Managing Partner of Catalio Capital Management. “We are delighted to support the company’s pipeline growth and advancement and look forward to being a strong partner for the future.”
“This financing strategically expands NextPoint’s group of high-quality investors and further transforms the company’s innovative pipeline to progress monotherapy treatments with a novel clinical biomarker,” commented Detlev Biniszkiewicz, PhD, Chairman of NextPoint’s Board of Directors. “We are excited to reshape immunotherapy into precision oncology and give hope to patients living with cancer.”
“This new round of financing underscores the support and confidence of our premier syndicate of investors, and we are well positioned to build upon our growing pipeline of multi-modal therapeutics targeting the novel HHLA2 pathway,” said Ivan Cheung, CEO of NextPoint. “We are advancing a diverse set of assets into clinical trials to exploit HHLA2’s role as both a novel immune checkpoint and a tumor- targeting mechanism. We are thrilled to pioneer a new class of monotherapies to treat both hot and cold tumors.”
About NextPoint Therapeutics
NextPoint is advancing the field of immuno-oncology through its leading scientific work on the novel HHLA2 axis, also known as B7-H7. Our innovative approach integrates foundational science with a defined clinical biomarker strategy to deliver a new class of monotherapies for patients who do not benefit from PD-1/L1 inhibitors. NextPoint is simultaneously advancing therapeutic approaches utilizing the unique upregulation of HHLA2 in cancer as an anchor for tumor-targeting therapeutic modalities.
Our team of proven drug developers is working closely with our renowned scientific founders to launch a new world of precision immuno-oncology and beyond. To learn more, visit nextpointtx.com.
Contacts
Media Contact
Chelsea Rule
MacDougall Advisors
1(781)235-3060
crule@macdougall.bio
The funding will advance two precision TYK2 inhibitors into the clinic for multiple sclerosis, psoriasis and other severe autoimmune and neurologic conditions.
New investors include Dementia Discovery Fund, Leaps by Bayer, and UPMC Enterprises.
such as Alzheimer’s Disease and Amyotrophic Lateral Sclerosis (ALS). Sudo is also developing a potential first and best-in-class topical TYK2 inhibitor for psoriasis and other immune-mediated dermatologic diseases.
About Sudo Biosciences
Sudo Biosciences is a biopharmaceutical company committed to designing and developing novel medicines to transform patients’ lives. The company’s programs target the tyrosine kinase 2 (TYK2) pseudokinase domain. TYK2 is a key mediator in cytokine signaling pathways that have been linked to a broad range of immune-mediated inflammatory conditions. The company’s pipeline of next generation TYK2 inhibitors includes a potential first- and best-in-class brain- penetrant candidate for the treatment of multiple sclerosis and neurodegenerative diseases with underlying neuroinflammation and a potential first- and best-in-class topical candidate for immune-mediated dermatologic diseases. Both candidates are anticipated to enter clinical trials in 2024. Sudo Biosciences is based in Carmel, IN, with operations across the US and UK. For more information, visit www.sudobio.com.
About Dementia Discovery Fund
The Dementia Discovery Fund (DDF), managed by SV Health Investors, is the world's largest family of specialized venture capital funds that invests exclusively in companies developing or enabling novel therapeutics for dementia. Dementias, including Alzheimer's Disease, are arguably the largest unmet medical need, with over 55m patients worldwide. With more than $500m raised for this strategy, and offices in London and Boston, DDF capitalizes on global investment opportunities to fulfill its mandate of delivering measurable impact and generating significant financial returns. Utilizing its network of venture partners, entrepreneurs, leading scientists, and strategic partners, DDF invests in and creates new biotech companies and provides thought leadership in the field. DDF is enabled by its limited partners including major pharmaceutical companies (Biogen, Bristol Myers Squibb, Eli Lilly and Co., GSK, Johnson & Johnson, Otsuka (Astex), Pfizer and Takeda), along with AARP, Aegon, Bill Gates, British Patient Capital, NFL Players Association, Quest Diagnostics, UnitedHealth Group, and the non-profits Alzheimer's Research UK and LifeArc. Learn more at www.ddf.vc.
About Leaps by Bayer
Leaps by Bayer, a unit of Bayer AG, leads impact investments into solutions to some of today's biggest challenges in health and agriculture. The investment portfolio includes more than 60 companies. They are all working on potentially breakthrough technologies to overcome some specific challenges such as, e.g., developing a sustainable protein supply, reducing the environmental impact of agriculture, preventing or curing cancer, and others. www.leaps.bayer.com.
About UPMC Enterprises
UPMC Enterprises is the innovation, commercialization and venture capital arm of UPMC, a $24 billion health care provider and insurer based in Pittsburgh. With an emphasis on translational sciences and digital solutions, UPMC Enterprises provides its portfolio companies and partners with capital, connections and resources to develop solutions to health care’s most complex problems. Working in close collaboration with innovators from UPMC and the University of Pittsburgh Schools of the Health Sciences, as well as others worldwide, UPMC Enterprises strives to accelerate science from the bench to the bedside and has committed to investing $1 billion in novel drugs, diagnostics and devices by 2024.
Contacts
Media
Kimberly Ha
KKH Advisors
917-291-5744
kimberly.ha@kkhadvisors.com
Source: Sudo Biosciences
BOSTON, MA, February 5, 2024 - NodThera, a leading clinical-stage biotech developing brain-penetrant NLRP3 inhibitors to treat chronic inflammatory diseases, today announces the appointment of Thomas Jaecklin, M.D., M.Sc., FMH as Chief Medical Officer (CMO), with effect from today.
With more than 20 years of experience across global large pharma, biotech and academic medicine, Dr. Jaecklin is an accomplished late-stage drug development leader. He brings deep expertise across multiple therapy areas, including neuroscience and inflammation, with a strong track record in the execution of integrated drug development programs, progressing clinical assets through to regulatory and commercial success.
Dr. Jaecklin most recently served as Vice President, Global Program Head of the Small Molecule Portfolio at the global biotechnology company Galapagos NV (Euronext & NASDAQ: GLPG), where his strategic guidance led to multiple successful regulatory submissions and approvals. Prior to this, he was Senior Vice President, Head of Clinical Development at global biopharmaceutical company Mirum Pharmaceuticals (NASDAQ: MIRM). At Mirum, he initiated and drove the spin-off of two rare disease assets from Shire to successful registration and oversaw the regulatory submission and approval of Livmarli® (maralixibat chloride) less than 3 years later. He also played a pivotal role in the company’s creation and build-up, assisting in its $120 million Series A Financing and subsequent IPO. Earlier in his career, Thomas served in key leadership roles in clinical and medical assessment, business development and market access at Novartis.
Dr. Jaecklin earned his M.D. from the University of Geneva, followed by a M.Sc. from the University of Toronto. He is Swiss board-certified in paediatrics, neonatology and critical care medicine.
Alan Watt, Chief Executive Officer of NodThera, said: “We are delighted to welcome Thomas to the NodThera team, with his wealth of sector experience and successful execution of drug development programs across multiple therapeutic areas. These will be critical to us delivering our vision of a paradigm shift in the treatment of disease through the selective modulation of the NLRP3 inflammasome.”
Dr. Thomas Jaecklin, newly appointed Chief Medical Officer of NodThera, added: “The NLRP3 inflammasome is one of the most exciting emerging areas of therapeutic science, and NodThera’s best-in-class molecules hold great potential to address the unmet medical need in multiple neurodegenerative and cardiometabolic diseases. With a wealth of excellent data and two ongoing Phase Ib/IIa studies, I am excited to be joining the Company at such a critical juncture and look forward to working with the leadership team and Board to advance our clinical programs.”
NodThera’s pioneering, biomarker-rich Phase Ib/IIa study investigating the potential of its lead candidate NT-0796 in Parkinson’s disease, announced in June 2023, has successfully dosed all patients and is on track to read out in Q1 2024. This readout will be closely followed in 2Q24 with results from the Company’s ongoing Phase Ib/IIa cardiovascular risk trial in an inflamed obese population, further reinforcing NodThera's leadership position in the field.
For more information about NodThera please contact:
NodThera
Tel: +44 (0) 1223 608130
Email: info@nodthera.com
ICR Consilium
Amber Fennell, David Daley, Sukaina Virji
Tel: +44 (0)20 3709 5700
Email: nodthera@consilium-comms.com
About NodThera
NodThera is a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases. Led by an experienced management team, NodThera is combining a deep understanding of NLRP3 inhibition, pharmaceutical neuroscience expertise and precision molecular chemistry. Its two lead clinical candidates are oral, small molecule NLRP3 inflammasome inhibitors, which have demonstrated differentiated, potentially best-in-class clinical profiles with significant anti-inflammatory effects and the ability to penetrate different areas of the brain, offering distinct opportunities to treat multiple indications. The Company is backed by top-tier investors including 5AM Ventures, Blue Owl Capital, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Boston, MA, with additional operations in Cambridge, UK and Seattle, WA. Learn more at www.nodthera.com or follow the Company on LinkedIn.
Accomplished leader brings track record of value creation with scientifically unique assets to drive the company’s next phase of growth toward clinical success
Cambridge, MA – February 1, 2024 – NextPoint Therapeutics, a clinical-stage biotechnology company developing a new class of precision oncology therapeutics targeting the novel HHLA2 pathway, today announced the appointment of Ivan Cheung as Chief Executive Officer (CEO) and to the Board of Directors.
Mr. Cheung brings 25 years of drug development, approval and product launch achievements, and organizational leadership experience to NextPoint, most recently serving as Eisai’s U.S. CEO. With his appointment, NextPoint aims to deliver breakthrough clinical outcomes from precision immuno-oncology therapeutics targeting the HHLA2 checkpoint axis and create additional modalities leveraging HHLA2 as a novel tumor antigen.
Mr. Cheung succeeds Detlev Biniszkiewicz, PhD, Managing Director at MPM BioImpact, who successfully led the company from its inception in 2018 and will continue to serve as the Chairman of the NextPoint Board of Directors. Under Dr. Biniszkiewicz’s leadership, NextPoint raised Series A and B funding and was transformed into a clinical-stage company.
“We are thrilled to welcome Ivan to NextPoint and his proven leadership, deep industry expertise and commitment to scientific innovation make him the ideal person to shepherd the company into this exciting next chapter,” said Dr. Biniszkiewicz. “Ivan’s leadership was instrumental in bringing the targeted anti-cancer therapy lenvatinib and the first disease-modifying therapy for Alzheimer’s disease lecanemab to reach patients worldwide. I extend my heartfelt gratitude to the dedicated team at NextPoint who have made my time here so rewarding and am honored to chair the esteemed Board and continue contributing to the company’s success and growth.”
Ivan Cheung, CEO of NextPoint, commented, “NextPoint exemplifies what is next in oncology innovation and I am pleased to join this stellar team to make a lasting impact in the lives of cancer patients and their families. My life-long passion is to create positive social impact and advance public health by means of breakthrough medications, and NextPoint is the ideal place to translate that goal into reality. I look forward to building upon the strong foundation that Detlev has created and work with our incredible team in this next phase of growth to establish NextPoint as a high-value biotechnology company.”
At Eisai, Mr. Cheung guided the significant growth of the North America business to over $1.5 billion in revenue. Under his leadership, lenvatinib was successively launched in a range of tumor types, and combination clinical trials with a PD-1 inhibitor were initiated. In addition, Mr. Cheung led the groundbreaking development and the first-ever full U.S. approval for lecanemab, a monoclonal antibody therapeutic to treat the underlying pathophysiology of early-stage Alzheimer’s disease. His stewardship also led to the first wide reimbursement access from the Centers for Medicare & Medicaid Services for this class of medicines. Prior to Eisai, he was at Booz Allen Hamilton where he provided strategic and operational advice to pharmaceutical and biotech companies. Mr. Cheung received an MBA from Harvard Business School and a BSE from Duke University.
About NextPoint Therapeutics
NextPoint is advancing the field of immuno-oncology through its leading scientific work on the novel HHLA2 checkpoint axis. Our innovative approach integrates foundational science with a defined clinical biomarker strategy to deliver a new class of monotherapies for patients who do not benefit from PD- 1/L1 inhibitors. NextPoint is simultaneously advancing therapeutic approaches utilizing the unique upregulation of HHLA2 in cancer as an anchor for tumor-targeting therapeutic modalities. Our team of proven drug developers is working closely with our renowned scientific founders to launch a new world of precision immuno-oncology and beyond. To learn more, visit nextpointtx.com.
Contacts
Media Contact
Chelsea Rule
MacDougall Advisors
1(781)235-3060
crule@macdougall.bio
New location in Leiden, The Netherlands will serve as hub for European operations and production of QurAlis’ products for clinical trials through commercialization
CAMBRIDGE, Mass., January 3, 2024 – QurAlis Corporation, a clinical-stage biotechnology company driving scientific breakthroughs into powerful precision medicines that will alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative diseases, today announced the opening of its European Union (EU) headquarters in Leiden, The Netherlands. This new location will serve as the hub for the Company’s European operations including the production of QurAlis’ products for its clinical trials through commercialization.
“The expansion of our operations into Europe represents a new chapter for QurAlis, building upon the tremendous momentum of our organization,” said Kasper Roet, Ph.D., CEO and co-founder of QurAlis. “In a short period of time, QurAlis has made significant progress with regulatory approvals for our clinical programs in the EU, Canada, and the UK. The successful completion of the quality systems inspection by the Dutch regulatory agency will allow us to directly leverage our world-class ASO manufacturing expertise and control our end-to-end production supply chain. With our new European headquarters, and the skilled talent network in Leiden and the region, we will further strengthen our position as we bring breakthrough precision medicines to patients with ALS, FTD, and other neurodegenerative diseases.”
QurAlis is currently advancing a pipeline with therapeutic candidates that target specific components of ALS pathology and defined ALS patient populations based on both disease-causing genetic mutations and clinical biomarkers. The company is focused on patients who have a loss of Kv7.2/7.3, patients who have loss of STATHMIN-2, as well as patients with loss of UNC13A and impairment in synaptic signaling. The QurAlis team is leveraging insights, platforms, and successes in ALS to collaborate and expand its pipeline to other neurodegenerative diseases, such as FTD. There are currently no cures for ALS or FTD. Limited therapeutic options are available for ALS and FTD patients who are in desperate need for effective therapies.
“QurAlis’ new location in Leiden is designed to not just meet our current needs, but also to scale with our business as we continue to grow,” said Hagen Cramer, Ph.D., chief technology officer of QurAlis. “Our new European headquarters will allow us to release ASOs and other products into the European market for our programs so that we can deliver innovative solutions and make a meaningful difference in patients’ lives.”
In addition to the new Leiden office, QurAlis’ global corporate headquarters are in Cambridge, Massachusetts.
About QurAlis Corporation
At QurAlis, we are neuro pioneers on a quest to cure. We work with a relentless pursuit of knowledge, a precise attention to craft, and an optimistic mindset to discover and develop effective precision medicines that will alter the trajectory of amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), and other neurodegenerative diseases. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a pipeline with therapeutic candidates that target specific components of ALS and FTD pathology and defined patient populations based on both disease-causing genetic mutation(s) and clinical biomarkers. For more information, please visit www.quralis.com or follow us on Twitter @QurAlisCo.
Media contact:
Kathy Vincent
kathy@kathyvincent.com
310-403-8951
- Funding to advance potential first- and best-in-class brain-penetrant TYK2 candidate in multiple sclerosis and potential first- and best-in-class topical dermal TYK2 candidate in psoriasis into the clinic.
- Top-tier life science investor syndicate co-led by Enavate Sciences and TPG, with participation from Sanofi Ventures, Surveyor Capital (a Citadel company), Monograph Capital, Eventide Asset Management, 50 South Capital and existing investors Frazier Life Sciences and Velosity Capital.
CARMEL, IN – December 20, 2023: Sudo Biosciences (“Sudo”), a biopharmaceutical company committed to designing and developing best-in-class precision TYK2 (tyrosine kinase 2) inhibitors, today announced the close of a $116 million Series B financing round co-led by Enavate Sciences and TPG, which is investing in the company through TPG Life Sciences Innovations and The Rise Fund, with participation from Sanofi Ventures, Surveyor Capital (a Citadel company), Monograph Capital, Eventide Asset Management, and 50 South Capita as well as existing investors Frazier Life Sciences and Velosity Capital. The Company has raised a total of $157 million funding since its founding in 2020.
The Series B funding will be used to advance two investigational TYK2 candidates into the clinic next year. Sudo’s CNS program is a potential first and best-in-class brain-penetrant TYK2 inhibitor that has the potential to significantly advance the treatment of both the relapsing and progressive forms of multiple sclerosis as well as neurodegenerative conditions such as Alzheimer’s disease and amyotrophic lateral sclerosis (ALS). Sudo is also developing a potential first-in-class topical TYK2 inhibitor for psoriasis and other immune-mediated dermatologic diseases.
“We are thankful for the support of our premier life science investors, which will allow us to advance our two development candidates into the clinic,” said Scott Byrd, CEO, Sudo Biosciences. “With this financing, we are well positioned to progress our pipeline of next generation TYK2 inhibitors and pursue our mission of improving care for the millions of people living with multiple sclerosis, psoriasis and other severe autoimmune and neurologic conditions.”
“We were attracted to Sudo by the excellent science, experienced management team, and clinical potential of its brain-penetrant and topical allosteric TYK2 inhibitors” said Edd Fleming, MD, Executive Vice President, Commercialization, Enavate Sciences. “Severe neurologic diseases such as progressive forms of MS, Alzheimer’s and ALS have limited treatment options, and we believe Sudo’s CNS program has the potential to address these unmet needs.”
“We are excited to partner with the Sudo team to unlock the potential therapeutic applications for TYK2 inhibition in neuroinflammation and autoimmune diseases,” said Shinichiro Fuse, PhD, Business Unit Partner with TPG Life Sciences Innovations. “The pre- clinical data with Sudo’s CNS and dermatology TYK2 programs are very promising and support TYK2’s potential as a target in these therapeutic areas.”
In conjunction with the financing, Edd Fleming, Shinichiro Fuse, and Chris Gagliardi, PhD, Principal at Sanofi Ventures, will join the Board of Directors.
About Sudo Biosciences
Sudo Biosciences is a biopharmaceutical company committed to designing and developing novel medicines to transform patients’ lives. The company’s programs target the tyrosine kinase 2 (TYK2) pseudokinase domain. TYK2 is a key mediator in cytokine signaling pathways that have been linked to a broad range of immune-mediated inflammatory conditions. The company’s pipeline of next generation TYK2 inhibitors includes a potential first- and best-in-class brain-penetrant candidate for the treatment of multiple sclerosis and neurodegenerative diseases with underlying neuroinflammation and a potential first- and best-in-class topical candidate for immune-mediated dermatologic diseases. Both candidates are anticipated to enter clinical trials in 2024. Sudo Biosciences is based in Carmel, IN, with operations across the US and UK. For more information, visit www.sudobio.com.
About Enavate Sciences
Enavate Sciences is a portfolio company of Patient Square Capital dedicated to supporting therapeutic companies advancing medicines and enabling technologies with transformative potential to address patient need. Through the application of capital support and operational experience, Enavate strives to enable and empower a diverse portfolio of therapeutics companies to accelerate innovation. To learn more about Enavate, please visit www.enavatesciences.com.
About TPG
TPG is a leading global alternative asset management firm, founded in San Francisco in 1992, with $212 billion1 of assets under management and investment and operational teams around the world. TPG invests across a broadly diversified set of strategies, including private equity, impact, credit, real estate, and market solutions, and our unique strategy is driven by collaboration, innovation, and inclusion. Our teams combine deep product and sector experience with broad capabilities and expertise to develop differentiated insights and add value for our fund investors, portfolio companies, management teams, and communities.
Media Contact
For Sudo Biosciences:
Kimberly Ha
KKH Advisors
917-291-5744
kimberly.ha@kkhadvisors.com
For Enavate Sciences:
Doug Allen / Zach Kouwe
Dukas Linden Public Relations
646-722-6530
Enavate@DLPR.com
For TPG:
Courtney Power / Julia Sottosanti
media@tpg.com
1 As of September 30, 2023, including AUM attributable to TPG Angelo Gordon on a pro forma basis.

- Research completed through collaboration with leading institutions produced the structure of LINE-1 RT using X-ray crystallography and cryo-electron microscopy, which can enable further rational drug design
- Publication outlines key LINE-1 RT mechanisms that activate the innate immune system, leading to disease-driving inflammatory response
- Findings further support ROME’s development of novel LINE-1 RT inhibitors for autoimmune disorders
December 14, 2023 11:00 AM Eastern Standard Time
BOSTON --(BUSINESS WIRE). ROME Therapeutics, a biotechnology company harnessing the power of the dark genome to develop breakthrough medicines for serious diseases, today announced a landmark publication in Nature revealing the first high- resolution structure of the LINE-1 reverse transcriptase (RT). The publication, "Structural analysis and inhibition of human LINE-1ORF2 protein reveals novel adaptations and functions", by Baldwin et al., also validates that LINE-1 RT activity in the cytoplasm is a key driver of the innate immune response and supports ROME’s approach to developing novel LINE-1 RT inhibitors for autoimmune disorders. The supporting research was co-led by ROME as part of a significant collaboration with more than a dozen leading academic groups and industry organizations.
“ROME is proud to be part of this historic research effort to solve the structure of LINE-1’s RT, insights which have already informed the structure-based drug design work which served as the foundation of our lead program,” said Rosana Kapeller, M.D., Ph.D., President, CEO and Co-founder of ROME. “The publication’s biochemical analyses also provide the first confirmatory evidence of the mechanisms by which LINE-1 RT leads to a disease-driving inflammatory response, which is the basis for ROME’s development of novel LINE-1 RT inhibitors to treat autoimmune disease. We believe that sharing these data with the scientific community will galvanize research into the dark genome’s role in disease and human health.”
Dr. Kapeller continued, “We would like to dedicate this publication to our colleague, Eric Baldwin, who was a visionary research scientist in structure-based drug discovery and was instrumental in solving the crystal structure of LINE-1 RT. This research is a testament to Eric’s work and to his lasting scientific legacy, which continues to drive the field forward.”
LINE-1 is a virus-like element that makes up about one-fifth of the human genome and encodes two proteins, one of which is ORF2 protein (ORF2p). ORF2p is responsible for LINE-1’s RT and endonuclease activities, which have been implicated in the pathophysiology of cancers, autoimmune diseases and neurodegeneration.
ROME Publishes Landmark Nature Paper Revealing First High- Resolution Structure of LINE-1 Reverse Transcriptase (RT) for Drug Discovery
Research completed through collaboration with leading institutions produced the structure of LINE-1 RT using X-ray crystallography and cryo-electron microscopy, which can enable further rational drug design Publication outlines key LINE-1 RT mechanisms that activate the innate immune system, leading to disease-driving inflammatory response Findings further support ROME’s development of novel LINE-1 RT inhibitors for autoimmune disorders
In the Nature publication, ROME and collaborators report, for the first time, X-ray crystallography and cryo-electron microscopy (cryo-EM) studies of the RT domain of ORF2p in multiple conformational states, revealing two novel folded domains that contribute to unique aspects of the LINE-1 lifecycle and insertion mechanisms. Importantly, the study also provides the first conclusive evidence that LINE-1 RT can activate the innate immune system by synthesizing RNA-DNA hybrids in the cytoplasm, triggering signaling via the cGAS/STING pathway, resulting in interferon production. This viral mimicry may explain how LINE-1 contributes to autoimmune and other inflammatory diseases.
“We’ve been dreaming about solving the 3D structure of LINE-1 ORF2 protein for more than 20 years,” said Jef Boeke, Ph.D., Sol and Judith Bergstein Director, Institute of System Genetics and Professor, Department of Biochemistry and Molecular Pharmacology at NYU’s Grossman School of Medicine and a member of ROME’s Scientific Advisory Board. “I am certain that these new structural insights into LINE-1 RT will open countless lines of new scientific inquiry, as we seek to leverage the understanding of how LINE-1 contributes to disease and how to interrupt its pathological activity with new treatment approaches.”
About LINE-1
Long Interspersed Nuclear Elements, or LINEs, are a ubiquitous family of transposable, virus-like elements embedded in the dark genome at the DNA level. A LINE can make new copies of itself through a reverse transcriptase mechanism. LINE-1 encodes several proteins, including a protein called ORF2, which contains both a reverse transcriptase (RT) domain that can reverse transcribe the LINE-1 RNA, making a new DNA copy, and an endonuclease domain, that can insert the DNA copy into a new genomic location. Activity of LINE-1 RT in the cytoplasm can result in cytosolic RNA-DNA hybrids triggering an innate immune response that can drive autoimmune disease pathology. Through rational structure-based drug design, ROME has designed novel, selective and potent inhibitors of LINE-1 RT to block this immune response, which are now in preclinical development for lupus and other autoimmune diseases.
About ROME
ROME Therapeutics is developing novel therapies for a range of serious diseases, including autoimmune disease, cancer and neurodegeneration, by illuminating the role of the dark genome — the vast genomic expanse beyond the traditional genes, which includes virus-like repetitive elements and non-coding sequences — in human health and disease. Leveraging the company’s unprecedented data sciences platform, ROME has built a deep pipeline of therapies targeting the dark genome. To lead this exploration, ROME has assembled a team of world-class leaders in drug discovery and development across immunology, oncology, chemistry and machine learning. ROME is based in Boston, Mass. For more information, please visit www.rometx.com.
Contacts
Investors
Monique Allaire
THRUST Strategic Communications
monique@thrustsc.com
Media
Lisa Raffensperger
Ten Bridge Communications
lisa@tenbridgecommunications.com

- Icosavax stockholders to receive $15.00 per share in cash at closing plus non-tradeable contingent value right (CVR) of up to $5.00 per share
- Representing a total equity value of up to $1.1 billion including the CVR
SEATTLE, Dec. 11, 2023 (GLOBE NEWSWIRE) -- Icosavax, Inc. (Nasdaq: ICVX) today announced it has entered into a definitive agreement pursuant to which AstraZeneca, through an acquisition subsidiary, will initiate a tender offer to acquire all of Icosavax’s outstanding shares for a price of $15.00 per share in cash at closing, plus a non-tradable contingent value right to receive up to $5.00 in cash, payable upon achievement of specified regulatory and net sales milestones.
The upfront cash portion of the consideration represents an equity value of approximately $838 million and a 43% premium over Icosavax’s closing market price on December 11, 2023, and a 73% premium to Icosavax’s volume-weighted average price for the preceding 60 trading days. Combined, the upfront and maximum potential contingent value payments represent, if achieved, an equity value of approximately $1.1 billion and a 91% premium over Icosavax’s closing market price on December 11, 2023, and a 130% premium to Icosavax’s volume-weighted average price for the preceding 60 trading days.
The closing of the tender offer is subject to certain conditions, including the tender of shares representing at least a majority of the total number of Icosavax’s outstanding shares, and other customary closing conditions and regulatory clearances. Upon the successful completion of the tender offer, a subsidiary of AstraZeneca will be merged with and into Icosavax and any remaining shares of common stock of Icosavax will be cancelled and converted into the right to receive the same consideration (including the contingent value right) per share payable in the tender offer. Subject to the satisfaction of the conditions in the merger agreement, the acquisition is expected to close in the first quarter of 2024.
Adam Simpson, Chief Executive Officer, Icosavax, said, “We are pleased to announce the proposed acquisition of Icosavax by AstraZeneca as we believe it offers the opportunity to accelerate, and expand access to, our potential first-in-class combination vaccine for older adults at risk from RSV and hMPV. We look forward to combining our skills and expertise in advancing the development of IVX-A12, with AstraZeneca’s decades of experience in RSV, resources, and capabilities in late-stage development.”
Iskra Reic, Executive Vice President, Vaccines & Immune Therapies, AstraZeneca, said: “This virus-like particle vaccine technology has the potential to transform prevention against severe infectious diseases, including RSV and hMPV. With the addition of Icosavax’s Phase III-ready lead asset to our late-stage pipeline, we will have a differentiated, advanced investigational vaccine, and a platform for further development of combination vaccines against respiratory viruses. This aligns with our strategy to deliver a portfolio of therapies to address high unmet needs in infectious diseases, and our ambition to protect the most vulnerable patients who have high risk of severe outcomes.”
Concurrent with this press release, Icosavax issued a press release announcing positive topline interim results for Icosavax’s Phase 2 study of IVX-A12, a combination virus like particle (VLP) vaccine candidate targeting both respiratory syncytial virus (RSV) and human metapneumovirus (hMPV). The press release can be found at www.icosavax.com.
Centerview Partners LLC is serving as exclusive financial advisor to Icosavax and Latham & Watkins LLP is serving as legal counsel.
About Icosavax
Icosavax is a biopharmaceutical company leveraging its innovative VLP platform technology to develop vaccines against infectious diseases, with an initial focus on life-threatening respiratory diseases and a vision for combination and pan-respiratory vaccines. Icosavax’s VLP platform incorporates antigen design capabilities and technology to enable multivalent, particle-based display of complex viral antigens, which it believes will induce broad, robust, and durable protection against the specific viruses targeted. Icosavax’s lead program is a combination vaccine candidate targeting respiratory syncytial virus (RSV) and human metapneumovirus (hMPV). Its pipeline includes additional candidates that provide optionality as potential components of future combination and pan-respiratory vaccines, including influenza and SARS-CoV-2. Icosavax was formed in 2017 to advance the breakthrough VLP technology from the Institute for Protein Design at the University of Washington with the goal to discover, develop, and commercialize vaccines against infectious diseases. Icosavax is located in Seattle.
Additional Information and Where to Find It
The tender offer described above has not yet commenced. This communication is not an offer to buy nor a solicitation of an offer to sell any securities of Icosavax, Inc. The solicitation and the offer to buy shares of Icosavax’s common stock will only be made pursuant to a tender offer statement on Schedule TO, including an offer to purchase, a letter of transmittal and other related materials, that AstraZeneca PLC, AstraZeneca Finance and Holdings Inc. and Isochrone Merger Sub Inc. (Merger Sub), a wholly owned indirect subsidiary of AstraZeneca PLC, intend to file with the Securities and Exchange Commission (SEC). In addition, Icosavax will file with the SEC a Solicitation/Recommendation Statement on Schedule 14D-9 with respect to the tender offer. Once filed, investors will be able to obtain a free copy of these materials and other documents filed by AstraZeneca, Merger Sub and Icosavax with the SEC at the website maintained by the SEC at www.sec.gov. Investors may also obtain, at no charge, any such documents filed with or furnished to the SEC by Icosavax under the “Investors & News” section of Icosavax’s website at www.icosavax.com.
INVESTORS AND SECURITY HOLDERS ARE ADVISED TO READ THESE DOCUMENTS WHEN THEY BECOME AVAILABLE, INCLUDING THE SOLICITATION/RECOMMENDATION STATEMENT OF ICOSAVAX AND ANY AMENDMENTS THERETO, AS WELL AS ANY OTHER DOCUMENTS RELATING TO THE TENDER OFFER AND THE MERGER THAT ARE FILED WITH THE SEC, CAREFULLY AND IN THEIR ENTIRETY PRIOR TO MAKING ANY DECISIONS WITH RESPECT TO WHETHER TO TENDER THEIR SHARES INTO THE TENDER OFFER BECAUSE THEY CONTAIN IMPORTANT INFORMATION, INCLUDING THE TERMS AND CONDITIONS OF THE TENDER OFFER.
Forward-Looking Statements
The statements included above that are not a description of historical facts are forward-looking statements. Words or phrases such as “believe,” “may,” “could,” “will,” “estimate,” “continue,” “anticipate,” “intend,” “seek,” “plan,” “expect,” “should,” “would” or similar expressions are intended to identify forward-looking statements. The forward-looking statements are based on the company’s current beliefs and expectations and include, but are not limited to: statements regarding the planned completion of the transactions contemplated by the Agreement and Plan of Merger, dated as of December 11, 2023 (the Merger Agreement), by and among Icosavax, AstraZeneca and Merger Sub and the timing thereof; expectations regarding the benefits sought to be achieved in the transactions; Icosavax’s expectations regarding the potential benefits and commercial potential of its vaccine candidates and technology platform; the ability to advance the company’s development programs and the potential to accelerate and expand access to IVX-A12 and other future vaccine candidates; and AstraZeneca’s strategic vision. Risks and uncertainties that could cause results to differ from expectations include: uncertainties as to the timing and completion of the tender offer and the merger; uncertainties as to the percentage of Icosavax stockholders tendering their shares in the tender offer; the possibility that competing offers will be made; the possibility that various closing conditions for the tender offer or the merger may not be satisfied or waived, including the failure to receive any required regulatory approvals from any applicable governmental entities (or any conditions, limitations or restrictions placed on such approvals); risks that the milestones related to the contingent value rights are not achieved; the effects of disruption caused by the transaction making it more difficult to maintain relationships with employees, collaborators, vendors and other business partners; risks related to diverting management’s attention from Icosavax’s ongoing business operations; the risk that stockholder litigation in connection with the transactions contemplated by the Merger Agreement may result in significant costs of defense, indemnification and liability; potential changes in AstraZeneca’s strategic vision; risks that results of a clinical trial at a particular time point may not predict future results; potential delays in the conduct of and receipt of data from clinical trials; unexpected adverse side effects or inadequate immunogenicity or efficacy of the company’s vaccine candidates; competing approaches and approved vaccines limiting the commercial value of the company’s vaccine candidates; regulatory developments in the United States and other countries; and other risks and uncertainties pertaining to Icosavax’s business, including the risks and uncertainties detailed in Icosavax’s public periodic filings with the SEC, as well as the tender offer materials to be filed by AstraZeneca and Merger Sub and the Solicitation/Recommendation Statement to be filed by Icosavax in connection with the tender offer.
You are cautioned not to place undue reliance on these forward-looking statements, which speak only as of the date hereof. All forward-looking statements are qualified in their entirety by this cautionary statement and Icosavax undertakes no obligation to revise or update these statements to reflect events or circumstances after the date hereof, except as required by law.
Media Contact:
Jessica Yingling, Ph.D.,
Little Dog Communications Inc.
jessica@litldog.com
858.344.8091
Investor Contact:
Laurence Watts
Gilmartin Group, LLC
laurence@gilmartinir.com
619.916.7620
- Financing led by Sanofi Ventures with participation from new investor Bpifrance (through its InnoBio 2 fund), and existing investors Khosla Ventures and Seventure Partners.
- Funding expected to generate proof of concept clinical data in patients.
Paris, FRANCE – December 5 2023 – Eligo Bioscience, a pioneering gene-editing company focused on addressing diseases driven by the expression of bacterial genes from the microbiome, has announced a successful $30 million Series B funding round, led by Sanofi Ventures. This infusion of capital, supported by new investor Bpifrance (through its InnoBio 2 fund), and existing backers Khosla Ventures and Seventure Partners (with Health For Life Capital™), propels Eligo towards becoming a clinical-stage biotech. Concurrent with this financing Laia Crespo, Ph.D., Partner at Sanofi Venture and Benoit Barteau, Investment Director at Bpifrance will join the board of directors.
“We are excited to lead this financing for Eligo and support the company as they move to the clinic.” said Laia Crespo, Ph.D., Partner at Sanofi Ventures. “We are impressed by the unique delivery and editing technologies that Eligo has developed, and we believe this will set the stage for Eligo to forge new paths in the application of in vivo gene-editing technologies.”
“We are delighted to welcome additional prominent healthcare investors to our existing syndicate. We feel this reflects the strong support for our vision and confirms the potential of Eligo to create a novel class of transformative genetic medicines.” said Xavier Duportet, PhD, Chief Executive Officer of Eligo Bioscience. “This is a defining time for Eligo as this funding puts us in a strong position to make a significant leap in treating diseases by editing the genetic makeup of the human microbiome.”
This funding is earmarked for accelerating the development of Eligo's flagship program, EB005, which targets moderate to severe acne vulgaris, an inflammatory disease that affects about 3% of the global population. The investment will fuel pre-IND and IND activities to achieve early human data readouts in a Ph1b/2a clinical trial. Additionally, it will facilitate Eligo’s expansion into other chronic diseases, including oncology.
Eligo stands at the forefront of a biotechnological revolution. By focusing on the in-vivo delivery of genetic cargoes to the microbiome, Eligo’s technology goes beyond traditional gene therapy and gene editing, expanding the range of addressable genetic targets. Through precise genetic modification within the human microbiome, this unique approach holds the promise of radically altering the course of numerous chronic and life-threatening diseases that are either triggered or driven by the expression of bacterial genes.
About Eligo Bioscience
Eligo Bioscience is the world leader in microbiome in-vivo gene editing and is advancing a highly differentiated pipeline of precision medicines to address unmet medical needs in immuno- inflammation, oncology, and infectious diseases driven by the expression of deleterious bacterial genes.
Eligo was founded by Luciano Marraffini (Professor at The Rockefeller University and cofounder of Intellia Therapeutics, Timothy Lu (Professor at MIT, and CEO at Senti Biosciences), Dr. David Bikard (Professor at Institut Pasteur) and Xavier Duportet. Eligo was named a Technology Pioneer by the World Economic Forum and received venture capital funding from Sanofi Ventures, Khosla Ventures, BPI France, and Seventure Partners.
For more information, please visit: eligo.bio Contact: xavier@eligo.bio
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi. Sanofi Ventures invests in early-stage biotech and digital health companies with innovative ideas and transformative new products and technologies of strategic interest to Sanofi. Among these areas are oncology, immunology, rare diseases, vaccines, potential cures in other core areas of Sanofi’s business footprint, and digital health solutions.
Find out more: www.sanofiventures.com
About Khosla Ventures
Khosla Ventures invests in companies that are bold, early and impactful. The firm was started in 2004 by Vinod Khosla, co-founder of Sun Microsystems, to provide venture assistance to entrepreneurs. Headquartered in Menlo Park, Calif., Khosla Ventures invests in a range of areas including AI, climate, sustainability, enterprise, consumer, fintech, digital health, medtech and diagnostics, therapeutics and frontier technology.
Visit our website: www.khoslaventures.com
About Bpifrance and InnoBio 2
Bpifrance is the French national investment bank: it finances businesses – at every stage of their development – through loans, guarantees, equity investments and export insurances. Bpifrance also provides extra financial services (training, consultancy) to help entrepreneurs meet their challenges (innovation, export).
InnoBio 2 is an investment fund dedicated to life sciences, managed by Bpifrance, which is also one of the LPs alongside Sanofi, Boehringer Ingelheim, Takeda, Ipsen, Servier, BMS, European Fund of Investment and Pasteur Mutualité. InnoBio 2, with €203 million, aims to invest in companies developing innovative products and services, close to or in early clinical development, with the objective of bringing them until the clinical proof of concept. InnoBio 2 takes minority equity stake in companies and can lead or co-lead the investment rounds.
For more information, please visit: www.bpifrance.com
About Seventure Partners
Seventure Partners is a long term equity investor that actively supports innovative companies aiming at generating positive impacts onHumankind, Society, Sustainability and the Planet.
With €950m net commitments under management as of the end of 2022, Seventure is a leading venture capital firm in Europe investing since 1997 in innovative businesses with high growth potential in 2 main areas: (i) Life sciences and (ii) Digital technologies. With Health for Life Capital™ funds (€160m and €250m commitments respectively in 2 vehicles) and its co-investment funds, Seventure is a worldwide leader in microbiome investments with more than 20 microbiome companies in its portfolio to date.
For more details: http://www.seventure.fr/en
- Fundraise led by Sofinnova Partners, F-Prime Capital, Digitalis Ventures and Cambridge Innovation Capital with participation from Sanofi Ventures and the University of Cambridge Venture Fund
- Unique transgenic mouse platform harnesses natural power of T cells to build a portfolio of first-in-class cancer medicines
15 November 2023; Cambridge, England – T-Therapeutics (“the Company”), a biotechnology company developing next-generation TCR therapeutics designed to reshape the clinical landscape for cancer patients, today announces it has raised £48 million ($59 million) in a Series A financing led by Sofinnova Partners, F-Prime Capital, Digitalis Ventures and Cambridge Innovation Capital (CIC) with participation from Sanofi Ventures and the University of Cambridge Venture Fund. The proceeds will be used to discover and develop novel T cell receptor (TCR) therapeutics for cancer indications as well as inflammatory disorders. Concurrent with the financing, Graziano Seghezzi (Sofinnova Partners), Nihal Sinha (F-Prime), Samuel Bjork (Digitalis) and Robert Tansley (CIC) will join the Company’s Board of Directors.
T-Therapeutics, which was spun out of the University of Cambridge, has developed a proprietary transgenic mouse platform, OpTiMus®, which creates an almost unlimited repertoire of ‘optimal’ TCRs as building blocks for pioneering therapies.
Initially, these treatments are being designed to recognise specific cancers and recruit the patient’s own T cells to eradicate the tumour. T-Therapeutics is building a portfolio of transformational TCR- based medicines for cancer, addressing the limitations of current TCR therapies which only apply to certain cancers and lack specificity, leading to significant side effects. T-Therapeutics will also develop medicines which address various auto-immune disorders.
The team at T-Therapeutics includes highly experienced antibody engineers and drug developers who were responsible for the creation of the Kymab and PetMedix antibody discovery platforms and pipelines among other notable discoveries, including at Adaptimmune and GSK. Of note, Kymab was acquired by Sanofi in 2021 for $1.45 billion and PetMedix was acquired by Zoetis, the world’s largest animal health company, in September this year.
Professor Allan Bradley, CEO of T-Therapeutics, commented: “We’re delighted to have raised this Series A with such high-quality investors whose amazing networks and shared vision will help us deliver highly differentiated TCR cancer therapies. TCR therapeutics are very much at the dawn of their potential. We intend to replicate the success of therapeutic antibodies but build on this in a new dimension, by using the targeting domains of TCR receptors to take advantage of their much greater specificity for cancer cells compared to normal cells. The same logic can be used to target immunosuppressive biologics to tissues impacted by autoimmune disorders.
“By engineering a mouse that makes human TCRs, we are able to discover anti-cancer TCRs that are quantitatively and qualitatively better than those that can currently be isolated from humans or using display technologies. Our OpTiMus® platform provides an unbeatable starting point, a vast repertoire of unique, fully human TCRs, with the properties to make them ideal to develop into drugs.
“We can also use the OpTiMus® mouse with our decades of mouse genome engineering experience to better understand immune responses to TCR-based therapies, and interpret responses to other immunotherapy interventions such as T-cell engagers, checkpoint inhibitors or future therapies.”
Graziano Seghezzi, Managing Partner at Sofinnova Partners, said: “Our investment in T-Therapeutics is a reflection of our conviction in both the exceptional team and the transformative technology they've brought forward. T-Therapeutics represents the kind of groundbreaking venture Sofinnova is deeply committed to, which has the potential to redefine healthcare. We are proud to be alongside Allan and the team as they pioneer a new era in cancer treatment.”
– ENDS –
Contact Us
T-Therapeutics
Allan Bradley
info@t-therapeutics.com
ICR Consilium
Amber Fennell, Sukaina Virji, Lucy Featherstone, Max Bennett
t-therapeutics@consilium-comms.com
About T-Therapeutics
T-Therapeutics is a next-generation T cell receptor (TCR) company spun off from the University of Cambridge. The company was created to harness the power of T cell biology, evolved over millions of years, to create safe and effective treatments for many cancers and autoimmune diseases. We combine world-leading expertise in mouse genome engineering, deep expertise in biopharmaceutical drug development, single cell genomics, machine-learning and structural biology, anchored in a culture of creativity and collaboration. We are developing ‘optimal’ TCR based therapeutics using our proprietary OpTiMus® platform, based on a fully humanized TCR mouse that provides an almost unlimited source of unique, antigen-specific human TCRs. These TCRs are directed at multiple target classes, many of which have never been worked on before. We are developing a pipeline of first-in- class drugs that will become transformative medicines, reshaping the clinical landscape for patients with cancer or autoimmune diseases.
About Sofinnova Partners
Sofinnova Partners is a leading European venture capital firm in life sciences, specializing in healthcare and sustainability. Based in Paris, London and Milan, the firm brings together a team of professionals from all over the world with strong scientific, medical and business expertise. Sofinnova Partners is a hands-on company builder across the entire value chain of life sciences investments, from seed to later-stage. The firm actively partners with ambitious entrepreneurs as a lead or cornerstone investor to develop transformative innovations that have the potential to positively impact our collective future.
Founded in 1972, Sofinnova Partners is a deeply established venture capital firm in Europe, with 50 years of experience backing over 500 companies and creating market leaders around the globe. Today, Sofinnova Partners has over €2.5 billion under management. For more information, please visit: sofinnovapartners.com.
About F-Prime Capital
F-Prime Capital is a leading global venture capital firm investing in healthcare and technology to solve the greatest challenges in healthcare and medicine while delivering on the conviction that everyone deserves a gold standard of care.
For over 50 years and with a portfolio of nearly 300 companies and counting spread across the Americas, Europe, and Asia, our independent venture capital group combines singular and deep expertise in life science, healthcare, and technology to identify and support founders with the next big ideas in therapeutics, medtech, and healthcare IT and services. With over $4.5 billion dollars under management, our team has created or co-created over 30 companies including Denali, Beam, Innovent, Orchard and has helped build many others including Blueprint Medicines, Iora Health, PatientPing, Devoted Health, Prime Medicine and Ultragenyx.
About Digitalis Ventures
Digitalis Ventures backs founders solving critical problems in health. The firm invests in early-stage companies across life sciences, health technology & services, and animal health with the goal of supporting them through multiple rounds of financing. Digitalis is headquartered in New York City.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi. Sanofi Ventures invests in early-stage biotech and digital health companies with innovative ideas and transformative new products and technologies of strategic interest to Sanofi. Among these areas are oncology, immunology, rare diseases, vaccines, potential cures in other core areas of Sanofi’s business footprint, and digital health solutions. Find out more: www.sanofiventures.com.
About Cambridge Innovation Capital
Cambridge Innovation Capital (CIC) is a leading venture investor backing and building category-leading deep tech and life sciences companies. CIC was founded to improve the success rate of businesses originating from the University of Cambridge and the broader Cambridge ecosystem, to encourage more academics and entrepreneurs from the area to build businesses. CIC currently manages in excess of £0.5 billion and has invested in around 40 companies. CIC is a preferred investor for the University of Cambridge, Europe’s top source of founders for venture-backed start-ups.
Cambridge Innovation Capital Manager Limited (FRN:918898) is authorised and regulated by the Financial Conduct Authority. For more information, please visit www.cic.vc or follow us on Twitter at @CIC_vc and LinkedIn.
About the University of Cambridge Venture Fund
Cambridge Enterprise is responsible for supporting the translation of University of Cambridge research to create social and economic impact with global significance. We do this by helping innovators, experts and entrepreneurs use commercial avenues to develop their ideas and expertise for the benefit of society, the economy, themselves and the University.
Liaising with organisations both locally and globally, we offer expert advice and support in commercialisation and social enterprise, including help with academic consultancy services, the protection, development and licensing of ideas, venture building and venture funding. Deeply embedded in the UK’s leading innovation and entrepreneurial ecosystem and part of the University of Cambridge, we have strong relationships with the University, industry, investors, innovators and visionaries.
Powerful and comprehensive health monitoring platform now offers FDA-cleared biomarkers for pulse and respiratory rate
BOSTON, Nov. 2, 2023 /PRNewswire/ -- Empatica, a digital health and AI company developing medical-grade wearables and digital biomarkers for health monitoring and diagnostics, today announced US Food and Drug Administration (FDA) 510(k) clearance for two new digital biomarkers for its Empatica Health Monitoring Platform: pulse and respiratory rate.
With the addition of pulse and respiratory rate, the Empatica Health Monitoring Platform now includes six FDA-cleared digital biomarkers, among the most offered for use in clinical trials. These are among the 128 digital measures supported by the platform, the largest offering available in a single solution, delivering confidence to a broad spectrum of health care professionals and researchers as they seek to validate treatments, better understand diseases, and innovate to improve health outcomes.
"We are delighted to announce the expansion of our Empatica Health Monitoring Platform to encompass two additional FDA-cleared biomarkers, pulse rate and respiratory rate," said Marisa Cruz, Chief Medical Officer at Empatica. "This clearance reflects our continued commitment to rigorous analytical and clinical validation of digital biomarkers for use in clinical research and patient care. The Empatica Health Monitoring Platform is a leading example of how reliable, accurate, and intuitive technology can support development of novel therapeutics and improve patient outcomes."
The Empatica Health Monitoring Platform is a full-stack remote health monitoring and data collection solution for research and healthcare professionals, built on data collected by the company's medical-grade, EmbracePlus wearable. In addition to the EmbracePlus wearable, the Empatica Health Monitoring Platform also includes Empatica's proprietary Care software suite, secure cloud infrastructure, and clinically validated digital biomarkers.
The Empatica Health Monitoring Platform received its initial FDA clearance in November 2022. Included within that clearance were clinically validated digital biomarkers used to monitor Electrodermal Activity, SpO2, skin temperature and movement during sleep. Beyond the FDA-cleared measures provided, the Empatica Health Monitoring Platform also offers access to raw data from the EmbracePlus sensors, and over 100 research-grade biomarkers, making it a highly capable and versatile measurement tool for today's clinicians and researchers.
To learn more about utilizing the Empatica Health Monitoring Platform, please visit us at https://www.empatica.com/talk-to-our-team/.
About Empatica
Empatica Inc is a pioneer in continuous, unobtrusive remote health monitoring driven by AI. Empatica's platform and technology are used by thousands of institutional partners for research purposes, in studies examining stress, sleep, epilepsy, migraine, depression, addiction, and other conditions. Its flagship medical wearable, EmbracePlus, has been developed with key partners including HHS, USAMRDC, and the NASA-funded TRISH.
Media Contact:
Steven Burk,
sburk@realchemistry.com
SOURCE Empatica
- THN391 was found to be safe and well-tolerated
- THN391 demonstrated a prolonged half-life and dose proportional Cmax levels
- The data will be presented October 25-27th at CTAD 2023
SACRAMENTO, Calif., Oct. 24, 2023 (GLOBE NEWSWIRE) -- Therini Bio, Inc., a biotech company focused on developing fibrin targeted therapies to treat inflammatory neurodegenerative and retinal diseases, today announced interim results from the Phase 1 trial of its lead candidate,
THN391, for the treatment of dementia. The data will be detailed in a poster presentation at the 16th Clinical Trials on Alzheimer’s Disease (CTAD) conference taking place from October 25-27, 2023, in Boston, MA.
Vascular dysfunction is a key driver in many neurodegenerative diseases, with fibrin playing a major role. Fibrin is a protein that is essential for blood clotting, but factors such as aging, vascular and rheumatological diseases and genetic risks, have shown fibrin to also cause chronic neuroinflammation and innate immune activation, resulting in severe retinal and neurological diseases, including Alzheimer’s. THN391 is a potential first-in-class, therapeutic, monoclonal antibody that is designed to target the inflammatory properties of fibrin without disrupting coagulation and protective innate immunity.
The Phase 1 trial evaluating THN391 consists of single ascending dose (SAD) and multiple ascending dose (MAD) portions, which are designed to study the safety and tolerability of THN391 in healthy subjects, as well as to collect pharmacokinetic and immunogenic measures.
At the data cut-off date, the first three cohorts (0.3 mg/kg, 1.0 mg/kg, 3.0 mg/kg) of eight subjects each (n=24) had received a single dose of THN391. No drug related adverse events were reported and the therapy was found to be well-tolerated. In addition, dose proportional Cmax levels were observed. Data from the first two cohorts, 0.3 mg/kg and 1.0 mg/kg, demonstrated a half-life of approximately 50 days in both groups. Data from the 3.0 mg/kg cohort continues to mature and additional SAD 10 mg/kg and MAD 3.0 mg/kg cohorts have been initiated.
“We are truly encouraged by the results of our promising early clinical data, which has correlated extremely well to our preclinical work,” said Jeffrey Stavenhagen, Ph.D., Chief Scientific Officer of Therini Bio. “The safety profile and extended half-life observed in these initial cohorts has emboldened us further to investigate a once-monthly or greater dosing schedule and implement a clinical plan that optimally drives development of THN391 for the treatment of dementia forward.
We believe that THN391’s ability to block fibrin-mediated neuroinflammation has the potential to change the lives of millions of people around the world.”
Presentation Details:
Theme: Beyond Amyloid and Tau
Title: Translation Studies and Clinical Development of THN391, a Novel Anti-Fibrin Antibody for the Treatment of Dementia
Poster Number: P200
Presenter: Jeffrey Stavenhagen, Ph.D., Chief Scientific Officer of Therini Bio
Date and Location: The poster will be on display from 7:30 a.m. ET on Wednesday, October 25, 2023, until 4:30 p.m. ET on Friday, October 27, 2023, in the Poster Hall at the Boston Park Plaza Hotel, and will also be available on the CTAD23 digital platform, www.ctad23.com, at 9:00 a.m. ET on Tuesday, October 24, 2023.
The poster will be accessible to the public on Saturday, October 28, 2023, at 10:00 a.m. ET at www.ctad-alzheimer.com and in the Publications section of www.therinibio.com.
About Therini Bio, Inc.
Therini Bio is a biotech company focused on developing fibrin-targeted therapies to treat inflammatory neurodegenerative and retinal diseases. The Company is developing a pipeline of potential first-in-class therapies targeting toxic fibrin accumulation, for diseases including Alzheimer’s disease (AD), multiple sclerosis (MS), as well as in a variety of retinal diseases, such as diabetic macular edema (DME) where destructive inflammation plays a role in the disease process. The foundational science was licensed based on technology discovered in Katerina Akassoglou, Ph.D. laboratories at the Gladstone Institutes at the University of California San Francisco (UCSF) and formerly the University of California San Diego (UCSD). Therini Bio’s top-tier syndicate of life sciences investors includes the Alzheimer’s Drug Discovery Foundation, Dementia Discovery Fund, Dolby Family Ventures, Eli Lilly and Company, Foundation for a Better World, MRL Ventures Fund, Sanofi Ventures and SV Health Investors’ Impact Medicine Fund. For more information, visit www.therinibio.com.
NIH Disclosure
Research reported in this release was supported by the NIA of the NIH under Award Number U01AG073125. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Contact
Leslie Schulze, CPA,
CGMA Chief Financial Officer
lschulze@therinibio.com
- Aberrant NLRP3 activation is recognised as a critical driver of cardiometabolic disease
- A Phase Ib/IIa trial of lead candidate NT-0796 will assess inflammatory biomarkers and
other cardiometabolic endpoints in an inflamed obese population - Sub-cohort will examine potential of NLRP3 modulation to reduce neuroinflammation as a
contributor to cardiometabolic disease in these patients
BOSTON, MA, October 16, 2023 - NodThera, a leading clinical-stage biotech developing brain- penetrant NLRP3 inhibitors to treat chronic inflammatory diseases, today announces that the first patients have been dosed in a Phase Ib/IIa clinical trial evaluating the potential of its lead candidate, NT-0796, to assess cardiometabolic biomarkers in obese patients with risk factors for atherosclerotic cardiovascular disease. This follows the U.S. Food and Drug Administration’s (FDA) ‘safe to proceed’ letter in response to the Company’s Investigational New Drug (IND) application.
The NLRP3 inflammasome is a highly validated drug target that plays a pivotal role in controlling inflammatory diseases. NLRP3 activation is recognised as a critical driver of cardiometabolic disease with strong data supporting a role in atherosclerosis, myocardial infarction and heart failure.
Emerging evidence also suggests that NLRP3 activation can drive neuroinflammation (reactive gliosis) leading to dysregulated storage of body fat, energy utilisation and obesity. NodThera’s trial of its brain- penetrant candidate NT-0796 will explore the potential of central NLRP3 inhibition in the brain to reduce gliosis and other consequences of obesity.
The randomised, double-blind, placebo-controlled Phase Ib/IIa trial will evaluate the pharmacokinetic and pharmacodynamic (PK/PD) profile of NT-0796 in inflamed obese patients over 28 days. Up to 60 patients will be enrolled and randomised into two cohorts receiving either NT-0796 or placebo. The study’s primary endpoint is the change in baseline to Day 28 of high-sensitivity C-reactive protein (CRP) levels, a key peripheral inflammatory marker and known predictor of risk of developing atherosclerotic cardiovascular (CV) disease. Secondary endpoints include multiple inflammatory and CV-risk specific biomarkers.
Alan Watt, Chief Executive Officer of NodThera, said: “Atherosclerosis is the leading cause of death in Western populations and inflammation is increasingly recognised as playing a major role in all stages of the disease. Modulation of the NLRP3 inflammasome consequently holds great potential for the development of novel treatment approaches. Our strategy at NodThera has been to develop small molecule NLRP3 inhibitors that penetrate both tissues and brain, not just for treating neurological disease but for treating the central components of peripheral disease that are caused by neurological dysfunction.
“Novel in-house preclinical findings have confirmed the importance of inhibiting brain NLRP3 in cardiometabolic disease models. In evaluating an obese population at high risk of cardiovascular disease we aim to assess the impact of NLPR3 inhibition on both peripheral inflammation and reactive gliosis, both of which contribute to cardiometabolic dysfunction.”
An earlier first-in-human study of NT-0796 has confirmed the candidate’s excellent PK/PD profile, brain penetration and anti-inflammatory effects in healthy subjects. A Phase Ib/IIa study in Parkinson’s disease patients is ongoing following NT-0796’s demonstration of a reduction of multiple neuroinflammatory and inflammatory biomarkers in plasma and cerebrospinal fluid (CSF) of elderly volunteers.
For more information about NodThera please contact:
NodThera
Tel: +44 (0) 1223 608130 Email: info@nodthera.com
ICR Consilium
Amber Fennell, David Daley, Sukaina Virji Tel: +44 (0)20 3709 5700
Email: nodthera@consilium-comms.com
About NodThera
NodThera is a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases. Led by an experienced management team, NodThera is combining a deep understanding of NLRP3 inhibition, pharmaceutical neuroscience expertise and precision molecular chemistry. Its two lead clinical candidates are oral, small molecule NLRP3 inflammasome inhibitors, which have demonstrated differentiated, potentially best-in-class clinical profiles with significant anti-inflammatory effects and the ability to penetrate different areas of the brain, offering distinct opportunities to treat multiple indications. The Company is backed by top-tier investors including 5AM Ventures, Cowen Healthcare Investments, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Boston, MA, with additional operations in Cambridge, UK and Seattle, WA. Learn more at www.nodthera.com or follow the Company on LinkedIn.
- New investment from EQT Life Sciences and OrbiMed with participation from existing investors
- MinervaX will hold financial reserves of more than €125 million, following the financing
- The financing supports the Company’s efforts to commence a Phase III clinical trial of its Maternal Vaccine against Group B Streptococcus
Copenhagen, Denmark, 11 October 2023 – MinervaX ApS, a privately held Danish biotechnology company developing a novel, prophylactic vaccine against Group B Streptococcus (GBS), has today announced the completion of a EUR 54 million upsized financing. The financing includes investment from new investors EQT Life Sciences and OrbiMed, with participation from existing investors Novo Holdings, Pureos Ventures, Sanofi Ventures, Trill Impact Ventures, Adjuvant Capital, Wellington Partners, Industrifonden, Sunstone LifeScience Ventures, and LF Invest. Vincent Brichard of EQT Life Sciences and Tal Zaks of OrbiMed will join the MinervaX Board of Directors.
GBS is a leading cause of life-threatening infections in newborns as well as adverse pregnancy outcomes such as preterm delivery and stillbirths. Current prophylactic measures provide insufficient protection, meaning there is an urgent need to accelerate the development of a GBS vaccine. A video describing the unmet medical need for a GBS vaccine and details of MinervaX’s novel GBS maternal vaccine can be found on the Company’s website, here: https://youtu.be/HvkRJBqsjjY.
The Company is currently progressing two Phase II clinical trials in 470 pregnant persons across Denmark, United Kingdom, Uganda and South Africa. Initial data from these clinical trials are highly positive and demonstrate that the vaccine has an acceptable safety profile, is highly immunogenic and gives rise to functionally active antibodies. Details of MinervaX’s clinical trials can be found at clinicaltrials.gov under the identifiers NCT04596878 and NCT05154578. In addition to pregnant persons, MinervaX is also pursuing Phase I development of its novel GBS vaccine in Older Adults, under identifier NCT05782179.
This financing will enable MinervaX to progress its novel GBS vaccine towards Phase III clinical trials in 2024.
Per Fischer, CEO of MinervaX, said: “The addition of EQT Life Sciences and OrbiMed to our existing investor consortium further strengthens the Company’s resolve to advance our novel GBS vaccine towards Phase III clinical trials in pregnant persons. It also provides additional validation and recognition of the acceptable safety profile and strong data demonstrated in the Phase II clinical trials. We are delighted to welcome Vincent Brichard and Tal Zaks to the board of directors, who will bring invaluable vaccine expertise as we continue to address the pressing need for the development of a novel vaccine to address the unmet medical burden of Group B Streptococcus.”
Vincent Brichard, Venture Partner EQT Life Sciences, commented: “EQT Life Sciences is thrilled to take an active part in the MinervaX prophylactic vaccine against GBS with the hope to save newborns’ lives. We are impressed by the clinical data achieved so far, the quality of the team and the near-term milestones enabling MinervaX to start a registration trial.”
Tal Zaks, Partner at OrbiMed, added: “We recognize the unmet need for better protection against GBS disease for vulnerable populations and the potential for MinervaX’s vaccine to provide best-in-class efficacy. I look forward to working with the MinervaX team to support the full development of this program.”
ENDS
For further information please contact:
MinervaX
Per Fischer | Chief Executive Officer
Email: pbf@minervax.com
Optimum Strategic Communications
Mary Clark / Stephen Adams / Zoe Bolt
Email: minervax@optimumcomms.com
Tel: +44 (0) 203 882 9621
Notes to Editors:
About MinervaX
MinervaX is a Danish biotechnology company, established in 2010 to develop a prophylactic vaccine against Group B Streptococcus (GBS), based on research from Lund University. MinervaX is developing a GBS vaccine for maternal immunization, and now also for vaccination of older adults, with Phase II data suggesting superior efficacy compared with other GBS vaccine candidates in development. The latter are based on traditional capsular polysaccharide (CPS) conjugate technology. By contrast, MinervaX’s vaccine is a protein- only vaccine based on fusions of highly immunogenic and protective protein domains from selected surface proteins of GBS (the Alpha-like protein family). Given the broad distribution of proteins contained in the vaccine on GBS strains globally, it is expected that MinervaX’s vaccine will confer protection against virtually 100% of all GBS isolates. www.minervax.com
About Group B Streptococcus (GBS)
GBS is responsible for nearly 50% of all life-threatening infections in newborns. At any given time, some 15- 25% of women are spontaneously colonized with GBS, and they run the risk of transmitting the bacteria to their child in the womb, during birth and/or during the first months of life. GBS colonization may lead to late abortions, premature delivery, or stillbirth and, in the newborn child, may result in sepsis, pneumonia or meningitis, all of which carry a significant risk of severe morbidity, long- term disability or death.
Currently, the only preventative strategy available involves the use of intravenously delivered prophylactic antibiotics which cannot comprehensively prevent GBS infection in utero and do not protect against late- onset infection in newborns. Not only is this approach expensive and logistically challenging, it fails to cover all, including the most severe cases in the US and Europe, and is rarely available in resource- limited settings. Finally, it carries the risk for increasing antibiotics resistance, a recognized worldwide health threat.
The development of a GBS vaccine is also endorsed by Group B Strep Support and Group B Strep International, and GBS has been prioritized by several public health organizations including the WHO. Both increased uptake of immunization among pregnant women and greater awareness of the implications of GBS suggest that a safe and effective vaccine targeting GBS would be well suited to address this unmet need.
About EQT Life Sciences
EQT Life Sciences was formed in 2022 following an integration of LSP, a leading European life sciences and healthcare venture capital firm, into the EQT platform. As LSP, the firm raised over EUR 3.0 billion (USD 3.5 billion) and supported the growth of more than 150 companies since it started to invest over 30 years ago.
With a dedicated team of highly experienced investment professionals, coming from backgrounds in medicine, science, business, and finance, EQT Life Sciences backs the smartest inventors who have ideas that could truly make a difference for patients.
More info: www.eqtgroup.com
Follow EQT on LinkedIn, Twitter, YouTube and Instagram
About OrbiMed
OrbiMed is a healthcare investment firm, with approximately $17 billion in assets under management. OrbiMed invests globally across the healthcare industry through a range of private equity funds, public equity funds, and royalty/credit funds. OrbiMed's team of over 100 professionals is based in New York City, San Francisco, Shanghai, Hong Kong, Mumbai, Herzliya, London and other key global markets.
More info: www.orbimed.com
- Total of $260 million in Series B funding includes an additional $50 million from existing and new investors, Bioluminescence Ventures and Solasta Ventures
Menlo Park, Calif. – September 19, 2023 – ReCode Therapeutics, a clinical-stage genetic medicines company using precision delivery to power the next wave of mRNA and gene correction therapeutics, today announced the closing of an extension to its Series B financing, raising an additional $50 million , and the appointment of Kouki Harasaki, Ph.D., founding and managing partner of Bioluminescence Ventures (BLV), to the company’s board of directors.
The company recently concluded a final extension to its Series B financing, raising an additional $50 million, for a total of $260 million in Series B funding.
● New investors in the extension include BLV and Solasta Ventures
● The new investor proceeds were supported with strong support from existing investors,
including OrbiMed Advisors, AyurMaya, an affiliate of Matrix Capital Management, Leaps by Bayer, Vida Ventures, MPM Capital, Pfizer Ventures, EcoR1 Capital, Sanofi Ventures and Amgen Ventures, among others
● Proceeds from the financing will be used to advance ReCode’s primary ciliary dyskinesia and cystic fibrosis clinical development programs and to expand the company’s proprietary Selective Organ Targeting (SORT) lipid nanoparticle (LNP) pipeline to include mRNA and gene correction therapeutics for central nervous system, lung, liver and musculoskeletal indications
Dr. Harasaki is founding and managing partner at BLV. He brings more than 25 years of biomedical science experience in multiple therapeutic areas across major health systems, research institutes, biopharmaceutical corporations, technology companies and venture capital firms. Prior to founding BLV, he was managing director at M12/Microsoft Ventures, where he led life science investments and helped develop Microsoft’s corporate strategy in the field. Before M12, Dr. Harasaki was a senior partner at Andreessen Horowitz.
“We are delighted to welcome Kouki to the board of directors and are confident his broad experience across a number of key areas such as drug discovery, strategy, finance and business development will be invaluable in guiding ReCode as it advances and expands it robust clinical development plans in a number of important genetic medicine indications,” said Shehnaaz Suliman, M.D., MBA, M.Phil., chief executive officer, ReCode Therapeutics. “We are excited with our progress to the clinic as we advance our SORT LNP delivery platform, the first technology to enable highly targeted delivery of genetic medicines to organs, tissues and cells including and beyond the liver.”
“I am excited to join the ReCode team at this important juncture in its development. At BLV, we are focused on funding next generation therapeutics platforms and developing first- and best-in-class programs. ReCode, with its cutting-edge genetic medicine platform, is well aligned with our mission,” said Dr. Harasaki. “I look forward to working with the board and the senior leadership team at ReCode
to advance the next wave of genetic medicines to address a wide range of medical needs not possible with current therapies.”
“Throughout 2023, we made tremendous progress entering the clinic, strengthening our financial position and building out our leadership team to support our genetic medicines clinical development programs. We are delighted with the continued high-level of interest in our novel approach to the targeted delivery of genetic medicines from premier venture investors. We remain focused on achieving important upcoming clinical milestones, including dosing the first patients in our Phase 1 trial of RCT1100 for primary ciliary dyskinesia and we are also on track to file a number of investigational new drug applications with global regulators for RCT2100, our cystic fibrosis candidate, later this year,” added Dr. Suliman.
About ReCode Therapeutics
ReCode Therapeutics is a clinical-stage genetic medicines company using precision delivery to power the next wave of mRNA and gene correction therapeutics. ReCode’s Selective Organ Targeting (SORT) lipid nanoparticle (LNP) platform enables highly precise and targeted delivery of genetic medicines directly to the organs and cells implicated in disease, enabling improved efficacy and potency. ReCode’s lead programs include RCT1100 for the treatment of primary ciliary dyskinesia caused by pathogenic mutations in the DNAI1 gene, and RCT2100 for the treatment of the 10-13 percent of cystic fibrosis patients who have Class I mutations in the CFTR gene and do not respond to currently approved CFTR modulators. RCT1100 and RCT2100 are inhaled disease-modifying mRNA-based therapies formulated using the SORT LNP delivery platform. ReCode is expanding its pipeline to develop potential therapies for other rare and common genetic diseases including musculoskeletal, central nervous system, liver and infectious disease indications.
ReCode’s SORT LNP platform was described by Nature as one of the “Seven Technologies to Watch in 2022” and the company was named among Fierce Biotech’s “Fierce 15” as one of the most promising early-stage biotechnology companies. ReCode has also been recognized by the San Francisco Business Times and Silicon Valley Business Journal as a Best Place to Work. For more information, visit www.recodetx.com and follow us on LinkedIn.
Investor Contact:
Anne Marie Fields
Managing Director
Stern IR
annemarie.fields@sternir.com
IR@recodetx.com
Media Contacts:
Erica Jefferson
SVP, Corporate Affairs ReCode Therapeutics
ejefferson@recodetx.com
650-629-7965
Tara Cooper
Founder and Principal
The Grace Communication Group
tara@gracegroup.us
650-303-7306
- Strategic investors Johnson & Johnson Innovation-JJDC Inc and Bristol Myers Squibb join Series B syndicate, alongside new and existing investors
- Funds will support early clinical development of LINE-1 reverse transcriptase inhibitor and further advancement of pipeline and platform
BOSTON – Sept. 12, 2023 – ROME Therapeutics, a biotechnology company harnessing the power of the dark genome to develop breakthrough medicines for serious diseases, today announced the completion of an oversubscribed $72 million Series B extension financing, bringing the total Series B amount raised to $149 million. The financing expands ROME’s investor syndicate to include new investors Johnson & Johnson Innovation-JJDC, Bristol Myers Squibb, Eurofarma Ventures, Luma Group, Mirae Asset Capital, and family offices Raycap and Sigmas Group. Existing investors ARCH Ventures, GV, Section 32, Sanofi Ventures, Andreessen Horowitz, Mass General Brigham Ventures, Casdin Capital, and Alexandria Venture Investments also participated in the round.
“We’re proud to have strong support from such high-quality investors, including strategic investment funds from four pharmaceutical companies whose participation demonstrates significant industry interest in the breakthrough potential of our lead development candidate in autoimmune disease and our platform architected to unlock the dark genome for drug discovery and development,” said Rosana Kapeller, M.D., Ph.D., President, Chief Executive Officer and Co-founder of ROME. “The capital from this raise enables us to progress our lead program into clinical trials and advance our pipeline and platform — collectively demonstrating the ability to translate our unique understanding of the dark genome, specifically the viral-like elements within it, into transformative therapies.”
ROME plans to use the funds raised in the Series B extension to advance its drug candidate, an inhibitor of LINE-1 reverse transcriptase (RT), through early clinical trials, including Phase 1 studies to evaluate safety and determine optimal dose, and additional studies designed to show proof of mechanism. LINE-1 RT is a viral-like protein encoded by the LINE-1 element, whose activity triggers innate immune responses that contribute to the development of autoimmune diseases. LINE-1 RT is expressed in diseased, but not healthy, cells and therefore LINE-1 RT inhibitors may block pathogenic inflammation without compromising response to infection.
ROME plans to develop the drug candidate for a number of serious autoimmune conditions, including lupus, in which LINE-1 is aberrantly expressed.
ROME also plans to continue advancing both its early pipeline and its proprietary data science platform that allows the company to identify functionally active repeat elements and assess their roles in disease. Using this platform, ROME is progressing several additional programs that are first-in-class therapeutic opportunities for autoimmune disease, cancer, and neurodegeneration. The platform is also informing ROME’s clinical trial design and providing insights to support patient selection in future clinical trials.
“The significant interest in this fundraise, particularly given the challenges of the current financing environment, is testament to the ROME team, science, and opportunities,” said Jeff Hatfield, Chair of ROME’s Board of Directors. “We’re delighted to have attracted both premier venture capital groups as well as industry-leading strategic pharma investment groups to join us in charting ROME’s next chapter as a clinical-stage company.”
About ROME Therapeutics
ROME Therapeutics is developing novel therapies for a range of serious diseases, including autoimmune disease, cancer, and neurodegeneration, by illuminating the role of the dark genome — the vast genomic expanse beyond the traditional genes, which includes viral-like repetitive elements and non-coding sequences — in human health and disease. Leveraging the company’s unprecedented data sciences platform, ROME has built a deep pipeline of therapies targeting the dark genome. To lead this exploration, ROME has assembled a team of world-class leaders in drug discovery and development across immunology, oncology, chemistry, and machine learning. ROME is based in Boston, Mass. For more information, please visit www.rometx.com.
Media Contact
Lisa Raffensperger
Ten Bridge Communications
lisa@tenbridgecommunications.com
Investor Contact
Monique Allaire
THRUST Strategic Communications
monique@thrustsc.com
- Exclusive multi-product agreement to develop and commercialize prescription digital therapeutics to treat substance use disorders.
- Collaboration to start with a preliminary exploration phase to define the product scope of CT-102 to treat Opioid Use Disorder (OUD).
- Click Therapeutics will receive upfront license and early development payments, and is eligible to receive additional regulatory and commercial milestones payments plus double-digit royalties on global sales, with the opportunity for a larger deal based on additional products.
September 07, 2023 06:55 AM Eastern Daylight Time
NEW YORK--(BUSINESS WIRE)--Click Therapeutics today announced the execution of a new collaboration agreement with Indivior for the development and commercialization of prescription digital therapeutics to treat substance use disorders, beginning with Opioid Use Disorder (OUD). Through a novel mobile application, CT-102, the collaboration aims to help close major gaps in OUD treatment, such as access to high-quality, personalized psychosocial treatment.
Designed to work alongside pharmacotherapy, CT-102 will combine evidence-based behavioral therapy with tailored neuromodulatory interventions using Click’s AI-enabled platform to deliver personalized care to each patient. This collaboration will build on Indivior’s expertise in the commercialization of innovative treatments for substance use disorders and Click Therapeutics’ substantial real-world experience engaging patients seeking help with addiction.
Click will lead development with an iterative, patient-centric and evidence-based approach that leverages the company’s end-to-end development capabilities and proprietary technology platform. This phased process is designed to ensure CT-102 will be launched as a best-in-class therapy supported by compelling evidence that will drive broad access and adoption. These efforts will start with a preliminary exploration phase to explore and define the product scope that will best match the needs of patients, providers and payers. More information will be available following completion of the exploration phase in 2024.
This is the fourth major co-development and commercialization collaboration to be announced that is based on Click Therapeutics’ clinically validated platform. Under the terms of the collaboration agreement, Click Therapeutics will be responsible for all technical and clinical development activities, as well as regulatory filings and technical services through commercialization. In return, Indivior will receive a global license to commercialize CT-102. Click Therapeutics will receive upfront license and early development payments, and is eligible to receive additional regulatory and commercial milestones plus double-digit royalties on global sales. In addition, there is an opportunity for a higher total deal value based on the potential development of additional products in the future.
About Click Therapeutics
Click Therapeutics, Inc., develops, validates, and commercializes software as prescription medical treatments for people with unmet medical needs. As a leading innovator of Digital Therapeutics™, Click delivers accessible, clinically proven, FDA-regulated prescription treatments to the smartphone in your hand. Click’s treatments are defined by a commitment to applying technical and scientific rigor and patient-centric design to the development process. This results in uniquely engaging experiences that achieve compelling clinical outcomes for patients seeking new treatment options. Click Therapeutics continuously expands and refines its platform with novel cognitive, behavioral and neuromodulatory mechanisms of action and advanced data-driven tools such as artificial intelligence and machine learning. The digital therapeutics under development on Click’s platform address diverse areas of therapeutic need, including indications in psychiatry, neurology, oncology, immunology, and cardiometabolic diseases. Consistently named a best place to work, Click fosters an inclusive, diverse workforce of innovators, clinicians, scientists, researchers, designers, technologists, engineers and more, united in a common mission to provide patients everywhere access to safe and effective prescription digital therapeutics.
For more information, visit www.clicktherapeutics.com and connect with us on LinkedIn.
Contacts
Investor
Daniel Busby
Media
Jonni Mills

- According to the analysis, MS and epilepsy patients experienced reduced annual healthcare costs of $6,280 and $4,600, respectively, on average after joining Medisafe
BOSTON, MA / ACCESSWIRE / September 6, 2023 / Medisafe, a leading digital health company specializing in medication engagement, released the results of a new retrospective, pre and post claims study of 207 patients with multiple sclerosis (MS) and epilepsy using Medisafe to manage their medication regimen and overall health.
The analysis showed an average annual cost reduction of $6,280 per MS patient and $4,600 per epilepsy patient using Medisafe to manage their therapeutic regimen.These cost reductions - which represent significant savings for patients and reduction in burden on the entire healthcare system - derived from reduced utilization of costly care services as a result of better medication management. Specifically, by using the Medisafe app to digitally manage their medication:
Patients with MS experienced a 34% reduction in total medical claims - including a 63% reduction in ER visits and 34% reduction in outpatient visits.
Patients with epilepsy experienced a 28% reduction in total medical claims - including a 36% reduction in ER visits and a 45% reduction in ambulance visits.
"By providing a digital health tool that helps patients manage their therapeutic regimens, their care is more well-managed, they are more engaged and their healthcare costs are reduced. As a result, providers are likely to achieve better reimbursement, payers are likely to spend less on patient care, and pharma is likely to see higher prescription rates," said Omri Shor, Medisafe Chief Executive Officer and co-founder. "This data both validates the role we are already playing in reducing the burden on the healthcare ecosystem, and reinforces our mission to expand that value to more patients over time."
The total estimated economic burden of MS and epilepsy combined is $113.4B, attributable partially to preventable utilization of high-cost acute care. For example, seizures or epilepsy account for 1% of annual emergency room visits, which averaged $1,150 in 2020 - 10x the rate of an urgent care or primary care provider (PCP). For patients with MS, the average excess cost of 3+ relapses requiring hospitalization is $15,079. Additionally, research shows ambulance rides average anywhere from $940 to $1,277 depending on the level of care needed and location - with some costing more and rising each year.
The analysis was conducted over a 14-month period on claims data from Medisafe users identified through patient match data from Komodo Health. Patients were first diagnosed between 1/1/21-1/1/22 and remained active on the Medisafe app into 2023. The study analyzed the volume of patient's medical claims during equal time periods prior to using the Medisafe app versus after.
Learn more about Medisafe's Digital Companions, Custom Branded Solutions, and Software Development Kit, and how together we can provide a virtual support system for your patients to help them stay engaged in their therapy, manage their care holistically, and live healthier lives.
About Medisafe
Medisafe's Connected Health Platform is the leading medication engagement platform that empowers patients to seamlessly manage their treatment journey while providing real-time intelligence and validated data to pharma partners, fortifying integration within the healthcare ecosystem. By combining advanced technology and behavior science, Medisafe reimagines the treatment journey to guide patients' specific journey needs and drive daily engagement. Its machine learning technology fuels the holistic patient engagement platform to personalize their support needs in a scalable fashion. By integrating existing patient support programs into its platform to extend capabilities, Medisafe is building a seamless future model of patient support and better health. Over 10M registered patients and caregivers rely on Medisafe's platform, delivering double digit results toward improving outcomes. The company manages over two billion medication doses via iOS and Android smartphones and tablets. With an average rating of 4.7 out of 5 stars and more than 400,000 user reviews, Medisafe helps to create more daily engagement than Facebook or Twitter applications. Medisafe is a HIPAA and GDPR compliant solution and ISO 27001:2013 and ISO 13845 certified.
Contact:
SOURCE: Medisafe
- Results showed TDP-43 pathology drives loss of synaptic UNC13A function; UNC13A is an essential regulator of neurotransmitter release at synapses
- UNC13A mis-splicing is critical genetic alteration in neurodegenerative diseases, occurring in 58 percent of all ALS patients and up to half of all frontotemporal dementia cases
CAMBRIDGE, Mass., September 6, 2023 – QurAlis Corporation, a clinical-stage biotechnology company developing breakthrough precision medicines for amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases with genetically validated targets, today announced it will present preclinical data showing that TDP-43 (TAR DNA-binding protein 43) pathology drives loss of synaptic UNC13A function in neurodegenerative diseases including ALS and frontotemporal dementia (FTD). Data also showed that an UNC13A splice-switching antisense oligonucleotide (ASO) prevented cryptic exon inclusion in UNC13A transcripts, increased UNC13A protein levels, and normalized localization of UNC13A protein at the synapse.
“UNC13A is a genetically validated target in ALS and FTD and our results showed that TDP-43 pathology drives loss of synaptic UNC13A function. At QurAlis, we hypothesize that ameliorating UNC13A mis- splicing using a splice-switching ASO can alleviate symptoms of ALS, FTD, and ALS/FTD spectrum disorder associated with synaptic dysfunction that underlies clinical manifestation and disease progression,” said Daniel Elbaum, Ph.D., chief scientific officer of QurAlis. “We recently launched our newest program that targets UNC13A mis-splicing and look forward to advancing this program along with our other programs targeting neurodegenerative diseases so that we can make a real difference in patients’ lives.”
QurAlis will present these data in a poster presentation at the 1st Biennial Conference on TDP-43 Function and Dysfunction in Disease in Trieste, Italy on Thursday, September 7, 2023.
About UNC13A
UNC13A is an essential regulator of neurotransmitter release at synapses. In ALS and FTD, the loss of TDP- 43 causes the mis-splicing of certain pre-mRNA transcripts resulting in expression of a cryptic exon- containing transcript that interferes with appropriate protein generation. An exon is a segment of a DNA or RNA molecule containing information coding for a protein or peptide sequence.
UNC13A is one of a number of pre-mRNAs that becomes mis-spliced due to loss of TDP-43 in disease. Fifty-eight percent of ALS patients and up to half of FTD patients carry a single nuclear polymorphism in the UNC13A gene or show TDP-43 pathology which greatly exacerbates UNC13A mis-splicing leading to loss of function of the UNC13A protein. There are currently no cures for ALS or FTD. Limited therapeutic options are available for ALS and FTD patients who are in desperate need for effective therapies.
Incorporating its proprietary FlexASOTM Splice Modulator Platform, QurAlis’ antisense oligonucleotides (ASOs) correct this mis-splicing, restore UNC13A protein production, and reduce cryptic exons that may contribute to disease progression.
The QurAlis FlexASO Splice Modulator Platform was developed to generate splice-switching ASOs with improved potency and an increased therapeutic index. In addition to UNC13A, QurAlis is currently exploring this ASO technology for multiple other disease targets.
About QurAlis Corporation
QurAlis is trailblazing the path to conquering amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases with genetically validated targets with next-generation precision medicines. QurAlis’ proprietary platforms and unique biomarkers enable the design and development of drugs that act directly on disease-causing genetic alterations. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a deep pipeline of antisense oligonucleotides and small molecule programs including addressing sub- forms of ALS that account for the majority of ALS patients. For more information, please visit www.quralis.com or follow us on Twitter @QurAlisCo.
Media contact:
Kathy Vincent
kathy@kathyvincent.com
310-403-8951
- Proprietary Ionic Liquids platform is advancing towards one of i2o’s main goals of producing oral biologics and peptides; an innovation that originated in Harvard’s labs under Dr. Samir Mitragotri
- Acquired multiple cardiometabolic assets including long-acting GLP-1s, Amylin, Glucagon and PYY that hold significant promise as combination therapies in diabetes, obesity and related indications
- Acquired ITCA 650 and the MediciTM Implant Platform, an investigational twice-yearly implantable GLP-1 for type 2 diabetes, recently granted a public hearing before an FDA Advisory Committee
BOSTON – August 28, 2023 – i2o Therapeutics, Inc. today announced the appointment of Kurt Graves as Chairman, President and CEO. He was previously Executive Chairman of the Board, a position he held since August 2021. Mr. Graves succeeds i2o co-founder and CEO Ravi Srinivasan, Ph.D., who is pursuing other leadership opportunities within the life sciences field.
While i2o has been actively advancing the ionic liquids technology in pre-clinical work on its lead GLP-1 asset, in parallel the company has completed a series of corporate and strategic transactions including:
- A $46 million Series A financing with top-tier biotech investors.
- An acquisition and integration of proprietary pipeline assets in the cardiometabolic space from Intarcia Therapeutics including long-acting GLP-1s, Amylin, Glucagon and PYY. The combination of long-acting GLP-1s and the other peptides noted hold significant potential to redefine the standards of care in diabetes, obesity, and related indications.
- An acquisition and integration of ITCA 650 and the Medici implant technology platform which is an investigational twice-yearly implantable GLP-1 for type 2 diabetes and was recently granted a public hearing on September 21 before an FDA Advisory Committee. The public hearing was granted by the Commissioner’s Office and FDA’s Chief Scientist.
- i2o has also entered into a new 4-year sponsored research and licensing agreement with the Mitragotri Lab at Harvard University to further strengthen its IP and leadership position in the Ionic Liquid space where additional ionic liquid uses and applications are being advanced.
A recent publication in the Lancet has estimated at the current pace that ~1.3 billion people will be living with diabetes by 2050 – a leading cause of death and disability worldwide. The authors concluded, “Diabetes will be a defining disease of this century,” and that “the world has underestimated the true scale and threat the disease poses…representing an urgent call to course correct.”
“The cardiometabolic assets and novel delivery platforms we have at i2o are important at a time when global policy and health officials are increasingly concerned by soaring diabetes and obesity prevalence rates and the massive implications that poor glucose and weight control will have on healthcare systems, societies, and global economies for decades to come,” said Kurt Graves, Chairman, President and CEO of i2o Therapeutics. “At i2o, our ultimate goal is to be an important part of the novel GLP-1 based combination products and innovative drug delivery solutions that are needed to raise the standards of care, address widespread unmet needs around poor control and poor adherence, and to help remove the barriers to access, affordability, and supply that are needed to help millions of patients.”
Mr. Graves has provided leadership to several highly innovative biotech and global pharmaceutical companies over the last 30 years. He has previously served as Chairman, President and CEO of Intarcia Therapeutics, former Chairman of Radius Health, former Executive Chairman of i2o Therapeutics, and as a Board member at Achillion Pharmaceuticals until it was acquired, and at Seres Health. He was also E&Y’s New England Entrepreneur of the Year in 2015. Previously, Mr. Graves served as EVP, Head of Corporate Development, Strategic Drug Development and Program Management and Head of Commercial at Vertex Pharmaceuticals. Prior to that, he was at Novartis Pharmaceuticals for nearly 10 years, most recently as the Global Head of the General and Specialty Medicines Business and the first Global Chief Marketing Officer for the Pharmaceuticals division. Earlier in his career, at Merck and Astra- Merck for nearly 10 years, he worked on multiple cardiovascular and GI medicines and led the GI Business Unit responsible for developing and commercializing Prilosec®, Nexium® and Prilosec OTC®.
About i2o Therapeutics
i2o Therapeutics is a private biotechnology company that was founded in 2019 to exploit the versatile properties of ionic liquids for producing oral biologics and peptides. i2o has also acquired wholly owned proprietary peptides with an intent to advance novel therapies for serious cardiometabolic diseases. As a complement to the oral ionic liquids approach, i2o also owns the rights to a sustained delivery system (small osmotic implants - MediciTM) that enable just twice-yearly maintenance dosing of chronic medications. With this scope of platform technologies and proprietary peptides, i2o’s initial aim is to form strategic partnerships and to advance meaningful innovations that achieve a new standard of care for patients suffering from T2DM, Obesity, and related diseases. Over time, i2o’s platforms will also be leveraged for other opportunities and collaborations.
Forward Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995, such as statements regarding the application and use of Ionic Liquids (ILs) technology in the development therapeutics for cardio-metabolic diseases, inflammatory diseases, and other partnered and target indications, the efficacy and safety of oral biologic products and combination peptides, patient delivery experience and compliance, the integration of acquired assets with our IL- platform, oral combination of a GLP-1 and other co-agonist peptides, our scientific collaborations with pharmaceutical companies and the potential outcomes of such collaborations, our planned preclinical and clinical programs and studies, the use of proceeds our from our Series A financing, and the potential commercialization of our products and partnership as compared to existing treatment options, among others. Statements in this press release that are not statements of historical fact are considered forward-looking statements, which are usually identified by the use of words such as “aims”, “anticipates,” “believes,” “continues,” “goal,” “could,” “estimates,” “scheduled,” “expects,” “intends,” “may,” “plans,” “potential,” “predicts,” “indicate,” “projects,” “seeks,” “should,” “will,” “strategy,” and variations of such words or similar expressions. Statements of past performance, efforts, or results of our preclinical and clinical trials, about which inferences or assumptions may be made, can also be forward-looking statements and are not indicative of future performance or results. Forward-looking statements are neither forecasts, promises nor guarantees, and are based on the current beliefs of i2o Therapeutics’ management as well as assumptions made by and information currently available to i2o Therapeutics. Such information may be limited or incomplete, and i2o Therapeutics’ statements should not be read to indicate that it has conducted a thorough inquiry into, or review of, all potentially available relevant information. Such statements reflect the current views of i2o Therapeutics with respect to future events and are subject to known and unknown risks, including business, regulatory, economic and competitive risks, uncertainties, contingencies and assumptions about i2o Therapeutics, including, without limitation, (i) the risks and uncertainties associated with the regulatory submission, review and approval process, (ii) the ability of i2o Therapeutics to continue its planned preclinical and clinical development of its development programs, and the timing and success of any such continued preclinical and clinical development and planned regulatory submissions, (iii) i2o Therapeutics’ ability to retain and hire key personnel, (iv) i2o Therapeutics’ ability to obtain additional financing to fund its operations and complete the development and commercialization of its various product candidates, (v) i2o Therapeutics’ ability to successfully commercialize its product candidates and uncertainties around regulatory reviews and approvals, (vi) i2o Therapeutics’ ability to scale its manufacturing and commercial supply operations for its product candidates and future approved products, (vii) i2o Therapeutics’ ability to obtain, maintain, protect and enforce patent protection and other proprietary rights for its product candidates and technologies, and (viii) the unknown future impact of the COVID-19 pandemic on certain clinical trials or their milestones and/or i2o Therapeutics’ business operations or operating expenses. i2oTherapeutics cautions you not to place undue reliance on any forward-looking statements, which speak only as of the date hereof. i2o Therapeutics does not undertake any duty to update any forward-looking statement or other information in this press release, except to the extent required by law.
Media contact:
Lauren Arnold
MacDougall Advisors
Larnold@macdougall.bio
- Financing co-led by Redmile Group and Bain Capital Life Sciences
- Proceeds to support completion of Phase 2/3 registrational trial of ABC008, a first-in-class anti- KLRG1 antibody for the treatment of inclusion body myositis as well as continued development of additional clinical programs
Newton, Massachusetts, August 17, 2023 – Abcuro, Inc., a clinical-stage biotechnology company developing therapies for the treatment of autoimmune diseases and cancer through precise modulation of cytotoxic T and NK cells, today announced the successful close of an oversubscribed $155 million Series B financing co-led by Redmile Group and Bain Capital Life Sciences.
New and existing investors also participated in the financing including RA Capital Management, Samsara BioCapital, Sanofi Ventures, New Leaf Ventures, Pontifax, funds managed by Tekla Capital Management, LLC, funds and accounts managed by BlackRock, Mass General Brigham Ventures, Eurofarma, and Soleus Capital.
“Support from such a strong group of investors will allow us to complete our development programs in diseases where there are few to no treatment options available,” said Alex Martin, Chief Executive Officer of Abcuro. “We are very motivated by the patients we serve and are excited by the clinical data we’ve seen to date. We’re committed to executing on our clinical trials including our registrational trial in inclusion body myositis.”
Abcuro will use the proceeds from the financing to complete a Phase 2/3 registrational clinical trial evaluating ABC008, a first-in-class monoclonal antibody targeting killer cell lectin like receptor G1 (KLRG1), for the treatment of inclusion body myositis (IBM). The Company will also focus on completing a Phase 1/2 clinical trial of ABC008 in T cell large granular lymphocytic leukemia (T-LGLL), as well as initiating a Phase 1/2 clinical trial in T and NK cell lymphomas.
“IBM, like other autoimmune diseases, is progressive and devastating for patients. Targeting the depletion of cytotoxic T cells that express KLRG1 with ABC008 is a novel approach that has generated exciting early data in patients with IBM,” said H. Jeffrey Wilkins, M.D., Chief Medical Officer of Abcuro. “These data are also supportive of using ABC008 in other diseases like T-LGLL in which cytotoxic T cells are pathogenic, and mature T and NK cell lymphomas in which KLRG1 expressing cells are malignant. We look forward to further advancing these programs in the clinic.”
About KLRG1
Killer cell lectin like receptor G1 (KLRG1) is an immune checkpoint cell surface receptor selectively expressed on late-differentiated effector memory (TEM) and effector (TEMRA) T cells. In autoimmune disease, highly cytotoxic T cells that drive disease progression express KLRG1. Conversely, in cancer, tumor cells expressing ligands that bind to the KLRG1 receptor inhibit effector T cells and natural killer (NK) cells and downregulate anti-tumor immunity. KLRG1 is, therefore, a compelling target in immune modulation in both autoimmune diseases and cancer as it enables the precise targeting of clinically relevant cytotoxic T and NK cells, while sparing naïve, central memory and regulatory T cells which are required to maintain normal immune system homeostasis.
About ABC008
ABC008 is a first-in-class anti-KLRG1 antibody capable of selectively depleting highly cytotoxic T cells, while sparing naïve, regulatory and central memory T cells. ABC008 has been designed to treat diseases mediated by highly cytotoxic T cells, including the autoimmune muscle disease inclusion body myositis (IBM), T cell large granular lymphocytic leukemia (T-LGLL), and mature T cell malignancies. ABC008 is currently being evaluated in a Phase 2/3 registrational trial for inclusion body myositis (IBM) and in a Phase 1/2 trial for T cell large granular lymphocytic leukemia. The US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have granted Orphan Designation to ABC008 for the treatment of IBM.
About Abcuro
Abcuro, based in Newton MA, is a clinical stage biotechnology company developing first-in-class immunotherapies for the treatment of autoimmune diseases and cancer through precise modulation of highly cytotoxic T and NK cells. The company is conducting two clinical trials in rare diseases: a Phase 2/3 registrational trial evaluating ABC008 in inclusion body myositis (IBM,) and a Phase 1/2 trial evaluating ABC008 in T cell large granular lymphocytic leukemia. For more information, visit us on LinkedIn and at abcuro.com.
Investor Contact:
Matt DeYoung
Argot Partners
abcuro@argotpartners.com
212-600-1902
Media Contact:
David Rosen
Argot Partners
david.rosen@argotpartners.com
212-600-1902
- Boston-based companies will collaborate on in vivo studies testing delivery modules harnessing transferrin and CD98 receptors -
BOSTON, Aug. 15, 2023 -- Chiesi Global Rare Diseases, a business unit of the Chiesi Group established to deliver innovative therapies and solutions for people living with rare diseases, today announced a co-development agreement with Aliada Therapeutics, a biotechnology company developing a novel blood-brain barrier (BBB)-crossing platform technology to address challenging disease areas with high unmet need.
The research collaboration will focus on multiple enzyme cargoes modified with Aliada's Modular Delivery (MODEL) platform, which harnesses endogenous brain endothelial cell transport mechanisms to efficiently move large molecule therapeutics across the BBB. Beyond enabling high therapeutic exposure in the brain, the MODEL platform demonstrates advantages over competing approaches with a broad design landscape that confers the ability to optimize therapeutics for both central nervous system (CNS) delivery and downstream functionality.
"Our commitment to the development of new treatment options for people living with lysosomal storage disorders (LSD) is global, as evidenced by recent regulatory approvals," said Giacomo Chiesi, head of Chiesi Global Rare Diseases. "Many LSDs have CNS involvement. With this collaboration, we are expanding our strategy and presence in BBB-crossing technologies and hope to leverage our know-how in LSDs to support the development of an effective and differentiated drug delivery platform. We are especially proud to advance this important research with Aliada, a partner with vast experience in neuro drug development and biologics delivery."
Founded in 2021, Aliada leverages its MODEL platform to research and develop differentiated large molecule therapeutics for patients with neurological disorders and systemic disorders with CNS involvement. Its leadership team brings deep experience and a successful track record across neuroscience and biologics discovery and development. Aliada is backed by leading life sciences investors, and plans to both advance proprietary programs and also collaborate with other companies to progress programs across multiple therapeutic areas.
"We are excited to partner with Chiesi to develop improved therapeutics for patients living with LSDs, who currently lack treatments that can readily access the brain," said Adam Rosenberg, Chief Executive Officer, Aliada Therapeutics. "We admire Chiesi's continued commitment to patients, exemplified by their two recent FDA approvals. This collaboration will allow Aliada to demonstrate the diverse capabilities of our MODEL platform, which enables us to efficiently transport a diverse range of therapeutic cargoes into the brain."
About Chiesi Global Rare Diseases
Chiesi Global Rare Diseases is a business unit of the Chiesi Group established to deliver innovative therapies and solutions for people affected by rare diseases. As a family business, Chiesi Group strives to create a world where it is common to have a therapy for all diseases and acts as a force for good, for society and the planet. The goal of the Global Rare Diseases unit is to ensure equal access so as many people as possible can experience their most fulfilling life. The unit collaborates with the rare disease community around the globe to bring voice to underserved people in the health care system.
For more information visit www.chiesirarediseases.com.
About Chiesi Group
Chiesi is an international, research-focused biopharmaceuticals group that develops and markets innovative therapeutic solutions in respiratory health, rare diseases, and specialty care. The company's mission is to improve people's quality of life and act responsibly towards both the community and the environment.
By changing its legal status to a Benefit Corporation in Italy, the US, and France, Chiesi's commitment to create shared value for society as a whole is legally binding and central to
company-wide decision-making. As a certified B Corp since 2019, we're part of a global community of businesses that meet high standards of social and environmental impact. The company aims to reach Net-Zero greenhouse gases (GHG) emissions by 2035.
With over 85 years of experience, Chiesi is headquartered in Parma (Italy), operates in 31 countries, and counts more than 6,500 employees. The Group's research and development centre in Parma works alongside 6 other important R&D hubs in France, the
US, Canada, China, the UK, and Sweden.
For further information please visit www.chiesi.com.
About Aliada Therapeutics
Aliada Therapeutics is a neuroscience-focused company working towards overcoming the delivery hurdle in large molecule drug development. Aliada is advancing a generation of CNS therapeutics using its novel BBB crossing platform technology, which can efficiently transport a diverse array of therapeutic cargoes into the brain, resulting in enhanced downstream effectiveness.
Chiesi Global Rare Diseases Media Contact
Adam Daley
Berry & Company Public Relations
Tel: +1 212 253 8881
Email: adaley@berrypr.com
Aliada Therapeutics Media Contact
Matt Crenson
Ten Bridge Communications
Tel: 917-640-7930
Email: mcrenson@tenbridgecommunications.com
PP-G-1247 V1.0
- Omada’s innovative member-to-provider virtual MSK program meets the industry’s top clinical standards with URAC accreditation for telehealth
August 01, 2023 - SAN FRANCISCO - Omada Health, a virtual-first healthcare provider specializing in chronic conditions, became the first virtual provider of musculoskeletal (MSK) care to earn the prestigious URAC telehealth accreditation. The Omada for MSK program is designed to improve timely access to high-quality MSK care. In the U.S., MSK conditions affect more than one in two adults and cost our healthcare system over $380 billion—more than diabetes, heart disease, or any other chronic condition.
“Earning this Telehealth Accreditation confirms that Omada Health is meeting the high clinical standards set by URAC,” said Omada Health Chief Medical Officer Carolyn Jasik, MD. “Achieving this status demonstrates our commitment to clinical best practices and patient safety. It also stakes our claim as the only provider in the digital MSK market that is able to pass this bar.”
URAC is the independent leader in promoting health care quality by setting high standards for clinical practice, consumer protections, performance measurement, operations infrastructure and risk management.
“Organizations that earn a Telehealth Accreditation demonstrate that they are operating at the highest standards possible in this continuously growing area of care delivery,” said URAC President and CEO Shawn Griffin, MD. “URAC’s Telehealth Accreditation shows Omada Health’s excellence at the important intersection of clinical best practices and technological safety and security.”
Many competing digital MSK care products are positioned as “preventive” or “consumer wellness programs,” as opposed to true clinical solutions. They often utilize remote monitoring with limited or no direct patient to physical therapist care. Omada for MSK, on the other hand, offers members end-to-end tele-physical therapy with licensed physical therapists; members can expect to meet directly with a licensed physical therapist for a telehealth visit within two days of signing up.
Omada for MSK, like Omada’s other programs, integrates high-quality, proactive behavioral health support. Omada’s licensed physical therapists consult with behavioral health specialists to help address behavioral health comorbidities that pose a significant risk to people dealing with MSK disorders. In fact, 17.8% of patients with MSK disorders also reported comorbid anxiety, while 11.5% reported depression. Addressing these issues together can drive cost savings of $460 per member per month.
“As a pioneer in the digital health provider industry, Omada continues to set high clinical standards in digital health care quality, safety, and accountability,” said Drew Contreras, PT, DPT, American Physical Therapy Association (APTA) vice president of clinical integration and innovation. “Obtaining clinically sound accreditations such as URAC’s Telehealth accreditation is in line with APTA’s longtime commitment to promoting digital health transparency,” Contreras added.
After earning URAC accreditation, Omada now holds a growing roster of nationally recognized accreditations and certifications across their diverse and complementary healthcare services. In 2021, Omada became the first fully-virtual healthcare provider to earn the National Committee for Quality Assurance’s (NCQA) Population Health Program (PHP) Accreditation. This year, Omada for Diabetes and its Diabetes+Hypertension programs earned renewed NCQA accreditation through 2026––the longest accreditation possible. Omada’s URAC accreditation now provides Omada clinical accreditation across conditions with a significant link, as 58% of people with diabetes also deal with MSK disorders.
“Earning URAC accreditation for Omada for MSK demonstrates our commitment to excellence and serves as the latest milestone that validates Omada’s integrated virtual care strategy across chronic conditions,” said Omada’s Senior Director of Clinical Services Todd Norwood, DPT. “We’re proud of our role as a digital health industry leader addressing musculoskeletal health and comorbid conditions, in a robust way that truly impacts the daily lives of our members.”
About URAC
Founded in 1990 as a non-profit organization, URAC is the independent leader in promoting health care quality and patient safety through renowned accreditation programs. URAC develops its evidence-based standards in collaboration with a wide array of stakeholders and industry experts. The company’s portfolio of accreditation and certification programs span the health care industry, addressing health care management and operations, pharmacies, telehealth, health plans, medical practices and more. URAC accreditation is a symbol of excellence for organizations to showcase their validated commitment to quality and accountability.
About Omada Health
Omada Health is a virtual-first healthcare provider that nurtures lifelong health, one day at a time. Our care teams implement clinically-validated behavior change protocols for individuals living with prediabetes, diabetes, hypertension, and musculoskeletal issues for consistent improvements that stack up. With more than a decade of experience and data, and 24 peer-reviewed publications that showcase our clinical and economic results, we both improve health outcomes and contain healthcare costs. Our scope exceeds 1,800 customers, including health plans, health systems, and employers ranging in size from small businesses to Fortune 500s.
Omada is the first virtual provider to join the Institute for Healthcare Improvement’s Leadership Alliance, reflecting our aim to complement primary care providers for the benefit of our members, and affirming our guarantee to every partner: Omada works different.
Contacts
|
|
BOSTON, MA, July 11, 2023 – NodThera, a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases, today announces positive, initial data from four subjects in the elderly volunteer stage of its Phase Ib/IIa study evaluating the effects of its lead candidate NT-0796 on inflammatory and disease-specific biomarkers in the blood and cerebrospinal fluid (CSF). Alan Watt, Chief Executive Officer of NodThera, said: "Taken together, these initial findings represent the first unambiguous demonstration of modulation of neuroinflammation in a human population with an NLRP3 inflammasome inhibitor. In designing our Parkinson’s disease study, we deliberately chose to measure the effects of NT-0796 in an elderly volunteer population as the first stage, since age is a clear factor in increased neuroinflammation. "Demonstrating such rapid decreases in just 7 days, across a broad range of neuroinflammatory biomarkers in the CSF, particularly NfL, is a striking result, as other drugs have required an extended timeframe of months or even years to show reduction of this biomarker. Our data provide clear validation of our strategy to take highly differentiated brain penetrant molecules into the clinic and justify our confidence in the potential of NT-0796 to treat diseases such as Parkinson’s disease and Alzheimer’s disease." Professor Paul Matthews, Head of the Department of Brain Sciences in the Faculty of Medicine of Imperial College London, said: "These data, while still very preliminary, provide promising evidence of the potential of NLRP3 inhibition to modulate the neuroinflammation associated with Parkinson’s disease. This is an exciting area. Development of molecules based on this concept could lead to a step change in the treatment landscape for neurodegenerative diseases more generally." Significant anti-inflammatory effects in both plasma and CSF Initial data from the ongoing study confirm earlier findings from the completed first-in-human and preclinical studies with NT-0796 showing excellent pharmacokinetics with a novel capsule formulation. Subjects in the study were cannulated and CSF-sampled on Day 1 (pre-dose) and Day 7 following daily NT-0796 dosing. CSF drug levels were confirmed as consistent with previous observations and a range of inflammatory CSF biomarkers demonstrated meaningful reductions. Neurofilament light chain (NfL), exclusively synthesised in the central nervous system (CNS), decreased by approximately 25% over 7 days in the most inflamed subject and by 13% on average. NfL is now recognised by the Food and Drug Administration (FDA) as a key biomarker of neuroaxonal damage and neurodegeneration. A full panel of cytokines, chemokines and adhesion molecules known to be associated with neuroinflammation were determined in the CSF, with the most inflamed individuals again demonstrating the most robust reductions. As previously observed, the most inflamed subjects at baseline showed the largest decreases in key peripheral inflammatory markers, C-reactive protein (CRP) and fibrinogen. Consistent reductions in circulating levels of unstimulated IL-1β, IL-18 and TNFα were also seen in subjects on Day 7 compared to Day 1. NodThera’s pioneering, biomarker-rich Phase Ib/IIa study in Parkinson’s disease, previously announced in June 2023, is currently recruiting into the patient arm of the study. This innovative clinical biomarker panel was designed using the preclinical profile of NT-0796 which demonstrated modulation of cytokines, chemokines and markers of gliosis relevant to neuroinflammatory disease. For more information about NodThera please contact: NodThera Tel: +44 (0) 1223 608130 Email: info@nodthera.com Consilium Strategic Communications Amber Fennell, David Daley, Sukaina Virji Tel: +44 (0)20 3709 5700 Email: nodthera@consilium-comms.com
About NodThera NodThera is a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases. Led by an experienced management team, NodThera is combining a deep understanding of NLRP3 inhibition, pharmaceutical neuroscience expertise and precision molecular chemistry. Its two lead clinical candidates are oral, small molecule NLRP3 inflammasome inhibitors, which have demonstrated differentiated, potentially best-in-class clinical profiles with significant anti-inflammatory effects and the ability to penetrate different areas of the brain, offering distinct opportunities to treat multiple indications. The Company is backed by top-tier investors including 5AM Ventures, Cowen Healthcare Investments, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Boston, MA, with additional operations in Cambridge, UK and Seattle, WA. Learn more at www.nodthera.com or follow the Company on LinkedIn. |
- Full enrolment and dosing commenced with its novel, prophylactic GBS vaccine in both healthy older adults and older adult population with co-morbidities
- First immunological read-outs are anticipated in Q4, 2023
Copenhagen, Denmark, 27 June 2023 – MinervaX ApS, a privately held Danish biotechnology company developing a novel, prophylactic vaccine against Group B Streptococcus (GBS), has today announced the completion of enrolment and initial dosing in its Phase 1, clinical vaccine trial in older adults. The trial is taking place at CEVAC (Centre for Vaccinology), Ghent University, Belgium.
GBS is most commonly associated with pregnant women and newborn babies. However, invasive GBS disease infections in the elderly population are continuously increasing. These can have devastating consequences – particularly if the person has a serious health condition such as diabetes mellitus, cancer, or a suppressed immune system.
To tackle this issue MinervaX has expanded the development pipeline of its novel GBS vaccine to include older adults, addressing the global burden and urgent need for the development of a vaccine to prevent and reduce deaths associated with GBS across the population. In pregnant women. MinervaX is currently progressing two Phase II clinical vaccine trials for the prevention of life-threatening infections in newborns. The trials are demonstrating that the vaccine has an acceptable safety profile, is highly immunogenic and gives rise to functionally active antibodies.
In April 2023, the Company commenced enrolment for its Phase I clinical vaccine trial in older adults. Enrolment and administration of the first dose to all participants is now complete. Details of MinervaX’s ongoing clinical trials can be found at clinicaltrials.gov under the identifiers NCT04596878, NCT05154578 and NCT05782179.
The Phase I vaccine trial will investigate the vaccine’s safety and immunogenicity in both healthy older adults and older adults with underlying medical conditions, i.e., diabetes and/or obesity, in an age range of 55 to 75. Two dose levels are being investigated: a lower dose level of 50 μg of fusion protein, which is also used in MinervaX’s two Phase II clinical trials in pregnant women, as well as a higher dose level of 125 μg of fusion protein. In addition, all older adult participants will receive three doses of the vaccine. The administration of one more jab than in the Phase II clinical vaccine trial in pregnant women, as well as the investigation of a higher dose level, takes into account that older adults – especially those with comorbidities, tend to exhibit weaker immune responses. All participants have now received one dose and further dosing is progressing smoothly with first immunological read-outs anticipated in Q4, 2023.
Lidia Oostvogels, Chief Medical Officer of MinervaX, said: “The smooth completion of enrolment and dose escalation provides an indication of the overall acceptable reactogenicity profile of our novel GBS vaccine and replicates the findings in our two ongoing Phase II trials in pregnant women. This allows us to accelerate the development of this potentially lifesaving vaccine to address the global unmet medical need. I would like to thank the participants of the trial and the team at CEVAC who is being instrumental throughout the trial, and I look forward to providing initial results in Q4, 2023.”
Prof. Isabel Leroux-Roels, Principal Investigator at CEVAC, commented: “We at CEVAC are very pleased to be contributing to this Phase I trial against this severe disease. Recruiting the many volunteers for this hugely important trial is a step forward to demonstrate that MinervaX’s novel vaccine works. We are very grateful to all the volunteers involved in the trial and will be following up with each participant accordingly. We are excited to see the data reported later this year.”
For further information please contact:
MinervaX
Per Fischer | Chief Executive Officer
Email: pbf@minervax.com
Optimum Strategic Communications
Mary Clark / Stephen Adams/ Zoe Bolt
Email: minervax@optimumcomms.com
Tel: +44 (0) 203 882 9621
Notes to Editors:
About MinervaX
MinervaX is a Danish biotechnology company, established in 2010 to develop a prophylactic vaccine against Group B Streptococcus (GBS), based on research from Lund University. MinervaX is developing a GBS vaccine for maternal immunization, and now also for vaccination of older adults, likely to have superior characteristics compared with other GBS vaccine candidates in development. The latter are based on traditional capsular polysaccharide (CPS) conjugate technology. By contrast, MinervaX’s vaccine is a protein- only vaccine based on fusions of highly immunogenic and protective protein domains from selected surface proteins of GBS (the Alpha-like protein family). Given the broad distribution of proteins contained in the vaccine on GBS strains globally, it is expected that MinervaX’s vaccine will confer protection against virtually 100% of all GBS isolates. www.minervax.com
About Group B Streptococcus (GBS)
Streptococcus agalactiae or Lancefield’s Group B Streptococcus (GBS) is a common commensal in humans, approximately 25% of all adults will be colonised with GBS at any given time. Invasive GBS disease is normally associated with infection in pregnant women and new-born babies; however, invasive GBS disease in adults has been increasing over the last 40 years. The older adult population (>65 years of age) and adults with underlying chronic health conditions (diabetes mellitus, cancer, immune suppression, obesity) are at particular risk of invasive GBS disease.
Group B Streptococcus disease in non-pregnant adults causes secondary and primary bacteraemia, septic arthritis, endocarditis, prosthetic joint infection, and necrotising myositis and fasciitis.
It is apparent that outside of pregnancy and the neonatal period, GBS infection results in high morbidity and mortality rates. There is no preventative treatment, cases are managed with antibiotics when an infection is diagnosed. There is a clear unmet medical need for a preventative vaccine that could provide protection to all adults but particularly to the older adult population or those at risk of infection due to underlying medical or demographic conditions. In addition, the incidence is increasing and will probably continue to increase with an increasing older adult population and an increase in the prevalence of obesity and type 2 diabetes around the world.
- Collaboration will leverage Unlearn’s AI-powered digital twins in QurAlis’ clinical trials for their leading ALS therapies
CAMBRIDGE, Mass., and SAN FRANCISCO – June 27, 2023 – QurAlis Corporation, a clinical-stage biotechnology company developing breakthrough precision medicines for amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases with genetically validated targets, and Unlearn, a pioneering technology company innovating machine learning to revolutionize medical research, today announced they have entered into a collaboration to accelerate and optimize QurAlis’ clinical program in ALS with Unlearn’s advanced generative artificial intelligence (AI) technology.
Unlearn develops digital twins of clinical trial patients that are predictions of individual health outcomes under the control treatment over time. Digital twins are employed in randomized controlled trials (RCTs) called TwinRCTs to run more efficient trials that produce regulatory-suitable evidence.
“Advances in machine learning and AI make it possible to enhance trial power to detect a positive result when one truly exists while controlling for Type-1 error and significantly shorten timelines without introducing bias into the study,” said Kasper Roet, Ph.D., founder and chief executive officer (CEO) of QurAlis. “We are excited to partner with Unlearn to help advance our clinical program with AI and other innovative technologies to generate evidence suitable for supporting regulatory decisions and help speed new, lifesaving precision medicines to patients with ALS and other neurodegenerative diseases.”
The collaboration aims to reduce variability and increase the study power in QurAlis’ clinical trials for QRL-201 and QRL-101, the Company’s lead product candidates in ALS. Unlearn’s patented machine learning models are trained on existing clinical data. After validation, they are used to generate digital twins from baseline data for each patient enrolled in a TwinRCT, regardless of their randomization assignment. Prognostic scores derived from digital twins are incorporated into the trial’s primary analysis to precisely estimate treatment effects and control for Type-1 error.
“By using machine learning to leverage the wealth of existing patient data from completed clinical trials, our technology significantly shortens typical timelines by months while generating evidence suitable for supporting regulatory decisions,” said Charles Fisher, Ph.D., founder and CEO of Unlearn. “The AI technology we’re developing today will revolutionize the future of clinical research.”
QRL-201 is a first-in-class therapeutic product candidate aiming to restore STATHMIN-2 (STMN2) expression in ALS patients. STATHMIN-2 is a well-validated protein important for neural repair and axonal stability, the expression of which is significantly decreased in nearly all ALS patients. QRL-201 rescues STMN2 loss of function in QurAlis ALS patient-derived motor neuron disease models in the presence of TDP-43 pathology. QRL-201 recently entered the clinic in the first-ever clinical trial to evaluate a therapy that rescues STMN2 in people with ALS (ANQUR; NCT05633459).
QRL-201 is the second program in QurAlis’ pipeline to enter the clinic recently. In December 2022, QurAlis announced the Company initiated dosing of QRL-101 in a first-in-human Phase 1 clinical trial (NCT05667779). QRL-101 is a first-in-class selective Kv7.2/7.3 ion channel opener for the treatment of hyperexcitability-induced disease progression in ALS.
About QurAlis Corporation
QurAlis is trailblazing the path to conquering amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases with genetically validated targets with next-generation precision medicines. QurAlis’ proprietary platforms and unique biomarkers enable the design and development of drugs that act directly on disease-causing genetic alterations. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a deep pipeline of antisense oligonucleotides and small molecule programs including addressing sub-forms of ALS that account for the majority of ALS patients. For more information, please visit www.quralis.com or follow us on Twitter @QurAlisCo.
About Unlearn
Unlearn is a San Francisco-based technology company pioneering AI-generated digital twins that forecast health outcomes for individual patients. Founded in 2017, Unlearn brings together a team of world-class innovators at the intersection of artificial intelligence, clinical, and regulatory science to transform the future of medicine through generative AI. Unlearn's technology is regulatory-qualified and used by leading global pharmaceutical companies to run AI-powered clinical trials that reach full enrollment faster and bring new treatments to patients sooner. For more information, please visit https://www.unlearn.ai or follow @UnlearnAI on Twitter, @unlearn-ai on LinkedIn.
Contacts:
For QurAlis:
Kathy Vincent
310-403-8951
For Unlearn:
Heather D’Angelo

- New Financing of $24 Million Fully Funds the Phase 2 Clinical Program for Lead Candidate VLX-1005
- Company Prepares to Evaluate VLX-1005 in Heparin-Induced Thrombocytopenia (HIT)
FREDERICK, Md., June 20, 2023 (GLOBE NEWSWIRE) -- Veralox Therapeutics, a clinical-stage biotechnology company developing a new class of therapies targeting the 12-lipoxygenase (12-LOX) pathway to address some of medicine’s most persistent and serious immune-mediated diseases, today announced the appointment of Jonathan Mow as the company’s new chief executive officer.
Mr. Mow’s appointment comes as Veralox secured $24 million in funding to advance VLX-1005 through a Phase 2a proof-of-concept study evaluating its impact on heparin-induced thromobcytopenia (HIT), a life-threatening rare disease caused by an aberrant immune response to heparin exposure. The investment round included new investors Pappas Capital and NYBC Ventures and existing investors Hatteras Venture Partners, Sanofi Ventures, JDRF T1D Fund and Genesys Capital, amongst others. In conjunction with the financing, the company welcomes Peter Young of Pappas Capital as a director and Meg Wood of NYBC Ventures as an observer.
“VLX-1005 has great promise to revolutionize the treatment of HIT and other immune-mediated diseases,” Mr. Young said. “I am thrilled to join the board at such an exciting time, and to be working with a leader of Jonathan’s caliber to move into later stages of clinical development.”
“This is an exciting time for Veralox as we head into our proof-of-concept Phase 2 study with VLX-1005 for HIT, a serious complication subsequent to heparin exposure that is accompanied by significant morbidity and mortality,” Mr. Mow said. “Our novel approach with 12-LOX inhibitors has great potential in this and other diseases and I would like to thank our investors for their financial support of our important mission and giving me the opportunity to lead this world-class effort and team.”
Mr. Mow brings more than 25 years of accomplishments in biotechnology management to Veralox, most recently serving as CEO of PhaseBio Pharmaceuticals. At PhaseBio, he led the company’s scientific and business transformations, guiding the company from early-stage research to Phase 3 development, and through a successful initial public offering in 2018.
Earlier in his career, Mr. Mow served as vice president, business development for Amylin Pharmaceuticals until its sale to Bristol-Myers Squibb in 2012; was co-founder and vice president, commercial and business development of Corus Pharma, Inc. until its acquisition by Gilead Sciences in 2006; and headed business development for PathoGenesis Corporation until its acquisition by Chiron Corporation in 2000. Mr. Mow has also held positions in marketing, marketing research and sales at Bristol-Myers Squibb, Wyeth/Lederle International and Syntex Laboratories. He holds a B.S. in mechanical engineering from University of California at Berkeley and M.B.A. from Carnegie Mellon University’s Tepper School of Business.
About Veralox Therapeutics
Veralox Therapeutics Inc. is the clinical leader in developing first-in-class therapeutics targeting 12-lipoxygenase, pioneering a new class of therapies that treat the underlying pathologies of serious immune-inflammatory diseases with unmet medical needs. The company’s lead candidate, VLX-1005, is in development for the treatment of patients with heparin-induced thrombocytopenia (HIT). VLX-1005 has orphan drug designation in the United States and has been awarded Fast Track Designation by the U.S. Food and Drug Administration. Second generation therapeutic products are under development for type 1 diabetes and other immune-mediated and inflammatory diseases. For more information, visit our website: https://veralox.com/.
Media Contact:
Lisa Guiterman
Scient PR
202-330-3431
- Oral, small molecule, clinical candidates NT-0249 and NT-0796 demonstrate differentiated, potentially best-in-class clinical profiles with significant anti-inflammatory effects
- Proven to penetrate different areas of the brain, providing opportunity to treat multiple indications
- Pioneering, biomarker-rich Parkinson’s disease study now underway with NT-0796
BOSTON, MA, June 20, 2023 – NodThera, a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases, today announces positive data from first-in-human studies of its lead therapeutic candidates, NT-0249 and NT-0796, and provides an update on the Company’s priority clinical development programme.
In the studies, both candidates were shown to clearly inhibit the NLRP3 inflammasome, a highly validated drug target that plays a pivotal role in controlling inflammatory diseases. The differentiated design characteristics of each candidate enabled them to penetrate different areas of the brain for optimal drug distribution in a range of NLRP3-driven diseases.
Brain penetration and anti-inflammatory effects across both clinical programmes
Data from the recently completed multiple-ascending dose (MAD) cohorts of NT-0249’s first-in-human study confirm a potentially best-in-class pharmacokinetic/ pharmacodynamic (PK/PD) profile, suitable for once-daily dosing. NT-0249 demonstrated significant anti-inflammatory effects in healthy volunteers, with reductions in key inflammatory biomarkers, C-reactive protein (CRP) and maintained fibrinogen, that were throughout treatment. Levels of NT-0249 measured in the cerebrospinal fluid (CSF) additionally demonstrated high levels of brain penetration.
Findings from the completed first-in-human study of NT-0796, initially disclosed in September 2022, also confirm an excellent PK/PD profile, brain penetration and anti-inflammatory effects in healthy volunteers.
Both candidates were well tolerated, treatment emergent effects were predominantly mild and there were no serious adverse events (SAEs).
Priority development programme underway in Parkinson’s disease
Development of NT-0796 is now progressing in a pioneering, biomarker-rich Phase Ib/IIa study in Parkinson’s disease. The study is exploring the candidate’s effect on inflammatory and disease-specific biomarkers in the blood and CSF using an innovative clinical biomarker panel, designed using the preclinical profile of NT-0796 on cytokines, chemokines and markers of microgliosis and astrogliosis relevant to NLRP3 inhibition.
The initial stage of the study, in healthy, elderly volunteers, is already underway, investigating a modified formulation of the drug candidate designed for use in the upcoming patient arm of the study.
Alan Watt, Chief Executive Officer of NodThera, said: “As the burden of non-communicable diseases continues to rise globally, targeting chronic low-grade inflammation, through selective modulation of the NLRP3 inflammasome, holds enormous potential for the treatment of these diseases. Our strategy to design highly differentiated and brain penetrant molecules, which
is delivering on the promise that NLRP3 inflammasome modulation can change the treatment paradigm for chronic peripheral and neurodegenerative diseases. These excellent clinical data from both clinical candidates reinforce our confidence that NodThera has the clinical tools to address these challenges.”
For more information about NodThera please contact:
NodThera
Tel: +44 (0) 1223 608130
Email: info@nodthera.com
Consilium Strategic Communications
Amber Fennell, David Daley, Suki Virji
Tel: +44 (0)20 3709 5700
Email: nodthera@consilium-comms.com
About NodThera
NodThera is a leading clinical-stage biotech developing brain-penetrant NLRP3 inflammasome inhibitors to treat chronic inflammatory diseases. Led by an experienced management team, NodThera is combining a deep understanding of NLRP3 inhibition, pharmaceutical neuroscience expertise and precision molecular chemistry. Its two lead clinical candidates are oral, small molecule NLRP3 inflammasome inhibitors, which have demonstrated differentiated, potentially best-in-class clinical profiles with significant anti-inflammatory effects and the ability to penetrate different areas of the brain, offering distinct opportunities to treat multiple indications. The Company is backed by top-tier investors including 5AM Ventures, Cowen Healthcare Investments, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Boston, MA, with additional operations in Cambridge, UK and Seattle, WA. Learn more at www.nodthera.com or follow the Company on LinkedIn.
- Company’s FlexASO programs targeting STATHMIN-2 and UNC13A showed three-fold potency, two-fold biodistribution improvement, improved safety, and greatly reduced off-target activity over standard ASOs
- QurAlis’ FlexASOTM Splice Modulator Platform designed to improve ASO performance, correct mis-splicing to restore synapse function, optimize distribution to deeper brain regions, and improve disease outcomes
- Data to be showcased in an oral presentation at 3rd Annual Oligonucleotides for CNS Summit
CAMBRIDGE, Mass., June 8, 2023 – QurAlis Corporation, a clinical-stage biotechnology company developing breakthrough precision medicines for amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases with genetically validated targets, today announced it will present new preclinical data showing the potential of the company’s antisense oligonucleotides (ASOs) generated from its proprietary FlexASOTM Splice Modulator Platform. Data from two of QurAlis’ FlexASO programs targeting rescue of STATHMIN-2 (STMN2) and UNC13A showed up to three-fold improvement over standard ASOs in both potency and biodistribution, and significantly reduced off-target effects, often from approximately 50 to 0, in human motor neurons. In addition, QurAlis’ FlexASOs were not associated with cytokine or chemokine production. Observations from both programs therefore indicate that FlexASOTM technology results in a higher therapeutic index than observed with traditional ASO.
“QurAlis’ FlexASO platform is based on unique and novel insights. We designed the platform to improve ASO performance, correct mis-splicing, and optimize distribution to deeper brain regions with the goal of identifying novel precision-medicine candidates for serious neurodegenerative diseases like sporadic ALS and frontotemporal dementia,” said Kasper Roet, Ph.D., CEO and co-founder of QurAlis. “These new preclinical data showed that our ASOs targeting restoration of STMN2 and UNC13A exhibited significantly higher potency, greater biodistribution, and reduced off-target activity over standard ASOs. We are excited by these promising data and look forward to continue advancing our programs so that we can fulfill our mission of bringing transformative disease-modifying therapies to patients with significant neurodegenerative diseases in desperate need of options.”
QurAlis will present these data in an oral presentation at the 3rd Annual Oligonucleotides for CNS Summit being held June 7-8, 2023 in Boston, MA. Daniel Elbaum, Ph.D., chief scientific officer of QurAlis, will present on June 8, 2023 at 8:00 AM ET, in a talk entitled, “Recent Advances in the Development of Splice Switching Oligonucleotides for CNS Diseases.”
The QurAlis FlexASO Splice Modulator Platform was developed to generate splice-switching ASOs with improved potency and an increased therapeutic index. In addition to STMN2 and UNC13A, QurAlis is currently exploring this ASO technology for multiple other disease targets.
About STATHMIN-2, TDP-43, and UNC13A
STATHMIN-2 (STMN2) is a well-validated protein important for neural repair and axonal stability, the expression of which is significantly decreased in nearly all ALS patients. Also known as SCG-10, STMN2 is a protein essential for the stabilization of microtubules which form an important component of the cytoskeleton of cells and axons. STMN2 is highly expressed in human motor neurons, the cells that primarily degenerate in patients suffering from ALS. In animal models, STMN2 deletion was found to cause axonal degeneration and loss of muscle innervation, which is the primary functional deficit that leads to paralysis in ALS patients.
Using human neuronal stem cell models from ALS patients, QurAlis co-founder and former Harvard professor Kevin Eggan, Ph.D., discovered in 2019 that the expression of STMN2 is regulated by TDP-43. The Eggan Lab showed that loss of normal TDP-43 function leads to a highly significant decrease in expression of STMN2 and an impairment in neuronal repair which could be rescued by restoring STMN2 levels. These results were published in Nature Neuroscience.
In addition to nearly all ALS patients, TDP-43 pathology is also associated with approximately 50 percent of patients with frontotemporal degeneration (FTD), the second most common form of dementia; about a third of Alzheimer’s Disease patients; and up to seven percent of Parkinson’s disease patients.
UNC13A is an essential regulator of neurotransmitter release at synapses. In ALS and FTD, TDP-43 accumulates in the cytoplasm and no longer maintains its normal function controlling RNA metabolism in the nucleus. Due to its loss, certain pre-mRNA transcripts are mis-spliced resulting in expression of a cryptic exon-containing transcript that interferes with appropriate protein generation. UNC13A is a pre- mRNA that is mis-spliced due to loss of TDP-43 in disease. Fifty-eight percent of ALS patients and up to half of FTD patients carry a single nuclear polymorphism in the UNC13A gene which greatly exacerbates UNC13A mis-splicing leading to loss of function of the UNC13A protein.
There are currently no cures for ALS or FTD. Limited therapeutic options are available for ALS and FTD patients who are in desperate need for effective therapies.
About QurAlis Corporation
QurAlis is trailblazing the path to conquering amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases with genetically validated targets with next-generation precision medicines. QurAlis’ proprietary platforms and unique biomarkers enable the design and development of drugs that act directly on disease-causing genetic alterations. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a deep pipeline of antisense oligonucleotides and small molecule programs including addressing sub- forms of ALS that account for the majority of ALS patients. For more information, please visit www.quralis.com or follow us on Twitter @QurAlisCo
Media contact:
Kathy Vincent
kathy@kathyvincent.com
310-403-8951
- EU CTA marks second regulatory clearance for Phase 1 ANQUR clinical trial of QRL-201 in ALS
- ANQUR is first-ever clinical trial to evaluate a therapy that rescues STATHMIN-2 expression in ALS patients
- QurAlis recently announced first patient dosed in Canada; enrollment completed in Cohort 1 portion of ANQUR
CAMBRIDGE, Mass., June 6, 2023 /PRNewswire/ -- QurAlis Corporation, a clinical-stage biotechnology company developing breakthrough precision medicines for amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases with genetically validated targets, today announced that the European review has been completed for the Clinical Trial Regulation (CTR) and each participating country has issued a notice of acceptance for QRL-201 for the potential treatment of ALS. QurAlis expects to initiate the Phase 1 ANQUR clinical trial of QRL-201 in participating European Union (EU) countries by the fourth quarter of 2023.
"This EU Clinical Trial Authorisation marks the second regulatory clearance for QRL-201 as we execute our global strategy focused on bringing breakthrough precision medicines to patients with ALS and other neurodegenerative diseases," said Kasper Roet, Ph.D., CEO and co-founder of QurAlis. "We believe QRL-201 has the potential to halt disease progression, which could transform the care and treatment of ALS. We are dedicated to ensuring efficient advancement of our ANQUR clinical trial so that we can fulfill our mission to make a meaningful difference in patients' lives."
QRL-201 is a first-in-class precision therapeutic product candidate aiming to restore STATHMIN-2 (STMN2) expression in ALS patients. ANQUR is the first-ever study to evaluate a therapy that rescues STMN2 expression in ALS patients. STMN2 is a well-validated protein important for neural repair and axonal stability and is the most significantly regulated gene by TDP-43 exclusively in humans. Its expression is significantly decreased in nearly all ALS patients and it is the most consistently decreased gene over all sporadic ALS patient data sets. QRL-201 rescues STMN2 loss of function in QurAlis ALS patient-derived motor neuron disease models in the presence of TDP-43 pathology.
The Clinical Trial Authorisation (CTA) in Europe is part of a global regulatory strategy established by QurAlis for the clinical development of QRL-201, which also includes a cleared CTA in Canada. QurAlis has completed enrolling patients in the Cohort 1 portion of the ANQUR clinical trial in Canada and recently announced the first patient has been dosed. Participating countries in the EU CTA include Germany, Belgium, the Netherlands, and Ireland.
About the ANQUR Clinical Trial
ANQUR (NCT05633459) is a first-in-human global, multi-center, randomized, double-blind, placebo-controlled multiple-ascending dose Phase 1 clinical trial designed to evaluate the safety, tolerability, and pharmacokinetics of QRL-201 versus placebo in patients with amyotrophic lateral sclerosis (ALS). The primary objective of the study is to determine the safety and tolerability of multiple doses of QRL-201 in people living with ALS. The ANQUR clinical trial is expected to include 64 study participants with ALS across sites in Canada, the European Union, U.S., and United Kingdom.
Visit www.clinicaltrials.gov for more information about the ANQUR study.
About STATHMIN-2 and TDP-43
STATHMIN-2 (STMN2) is a well-validated protein important for neural repair and axonal stability, the expression of which is significantly decreased in nearly all ALS patients. Also known as SCG-10, STMN2 is a protein essential for the stabilization of microtubules which form an important component of the cytoskeleton of cells and axons. STMN2 is highly expressed in human motor neurons, the cells that primarily degenerate in patients suffering from ALS. In animal models, STMN2 deletion was found to cause axonal degeneration and loss of muscle innervation, which is the primary functional deficit that leads to paralysis in ALS patients.
Using human neuronal stem cell models from ALS patients, QurAlis co-founder and former Harvard professor Kevin Eggan, Ph.D., discovered in 2019 that the expression of STMN2 is regulated by TDP-43. The Eggan Lab showed that loss of normal TDP-43 function leads to a highly significant decrease in expression of STMN2 and an impairment in neuronal repair which could be rescued by restoring STMN2 levels. These results were published in Nature Neuroscience.
In addition to nearly all ALS patients, TDP-43 pathology is also associated with approximately 50 percent of patients with frontotemporal degeneration (FTD), the second most common form of dementia; about a third of Alzheimer's Disease patients; and up to seven percent of Parkinson's disease patients.
There are currently no cures for ALS or FTD. Limited therapeutic options are available for ALS and FTD patients who are in desperate need for effective therapies.
About QurAlis Corporation
QurAlis is trailblazing the path to conquering amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases with genetically validated targets with next-generation precision medicines. QurAlis' proprietary platforms and unique biomarkers enable the design and development of drugs that act directly on disease-causing genetic alterations. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a deep pipeline of antisense oligonucleotides and small molecule programs including addressing sub-forms of ALS that account for the majority of ALS patients. For more information, please visit www.quralis.com or follow us on Twitter @QurAlisCo.
SOURCE QurAlis
- Industry Veteran Frank D. Lee Appointed as Chairperson of the Board
SOUTH SAN FRANCISCO – May 15, 2023 - Therini Bio, Inc., a biotech company developing fibrin-targeted therapies to treat inflammatory neurodegenerative and retinal diseases, today announced the initiation of first-in-human dosing for a Phase 1 trial of its lead asset THN391, a novel fibrin-targeting therapy for Alzheimer’s disease.
THN391 binds to the inflammation-driving component of fibrin that is known to activate pathological immune responses in neurodegenerative and ophthalmologic diseases. Importantly, based on preclinical studies to date, targeting this region does not impact or diminish fibrin’s critical role in blood clotting and coagulation. Key safety and proof of mechanism clinical data is expected by the end of 2024.
"Initiating first-in-human dosing for THN391 is a significant milestone for Therini Bio, and we are thrilled to be part of their journey to advance novel therapies for Alzheimer's disease. The team has made significant progress in a short period of time, and we look forward to our continued collaboration to advance THN391 for patients,” said Olga Danilchanka, Principal, MRL Ventures Fund, the therapeutics-focused corporate venture fund of Merck & Co. Inc.
“We’re excited about the novel approach that Therini Bio is taking towards treating Alzheimer’s disease and other inflammatory neurodegenerative and retinal diseases. The team is developing transformational new medicines, and we look forward to supporting their mission of developing potential first-in-class therapies targeting toxic fibrin accumulation for areas of significant unmet patient need,” said Laurence Barker, Partner, Dementia Discovery Fund (DDF).
As Therini Bio enters a new era of growth as a clinical-stage company, the Board of Directors has appointed industry veteran Frank D. Lee to Chairperson of the Board at Therini Bio. Frank was most recently President and CEO of Forma Therapeutics until its acquisition by Novo Nordisk for $1.1B in 2022. He brings over 25 years’ global experience in product development and commercial leadership across a wide range of therapeutic areas within the biotech and pharmaceutical industry. He was also Senior Vice President, Global Product Strategy and Therapeutic Area Head for the Immunology, Ophthalmology and Infectious Diseases at Genentech, a member of the Roche Group, where he was responsible for driving development and commercial strategy for a broad portfolio of molecules in development and for global in-line product sales of more than $11B.
Frank’s 13-year career path at Genentech included leadership positions of increasing scope and responsibility for delivering transformative medicines to patients. Prior to joining Genentech, Frank spent approximately 13 years across Novartis, Janssen and Eli Lilly in engineering, manufacturing, sales/marketing and business development. Frank received a bachelor’s degree in Chemical Engineering from Vanderbilt University and an MBA in marketing and finance from the Wharton Graduate School of Business. He currently services as chairman of the board of Catamaran Bio and as an independent board member of Bolt Bio. He previously served on the board of directors of the Genentech Foundation.
"We are thrilled to announce the initiation of first-in-human dosing for THN391, our novel fibrin-targeting therapy for Alzheimer's disease. This is a major milestone for the Company, and as part of our continued growth, we are excited to welcome Frank as Chairperson of the Board at Therini Bio. Frank's extensive experience in product development and commercial leadership will be invaluable as we enter this new era as a clinical-stage company," said Michael Quigley, Ph.D., President and CEO of Therini Bio.
"I am honored to be joining Therini Bio as Chairperson of the Board, as the Company advances its first clinical candidate THN391 as a novel fibrin-targeting therapy for Alzheimer's disease,” said Frank D. Lee, Chairperson of the Board, Therini Bio. “I look forward to working alongside the Therini Bio team to help advance this candidate and its pipeline of fibrin-targeted therapies. Therini Bio has assembled a talented group of experienced pharma and biotech veterans with a deep commitment to improving patient outcomes, and I am thrilled to be a part of this effort."
“Sanofi Ventures is passionate about supporting innovative companies that are developing breakthrough therapies for patients in need. We are thrilled to have Frank join us as Chairperson of the Board, as Therini Bio advances their novel fibrin-targeting therapy for Alzheimer's disease and retinal diseases. We look forward to working with the team throughout development in both indications,” said Laia Crespo, Partner, Sanofi Ventures.
About Therini Bio, Inc.
Therini Bio is developing fibrin-targeted therapies to treat inflammatory neurodegenerative and retinal diseases. The Company is developing a pipeline of potential first-in-class therapies targeting toxic fibrin accumulation, for diseases including Alzheimer's disease (AD), multiple sclerosis (MS), as well as in a variety of retinal diseases, such as diabetic macular edema (DME) where destructive inflammation plays a role in the disease process. The foundational science was licensed based on technology discovered in Katerina Akassoglou, Ph.D. laboratories at the Gladstone Institutes at the University of California San Francisco (UCSF) and formerly the University of California San Diego (UCSD). For more information, visit www.therinibio.com.
Media Contact
Kimberly Ha
KKH Advisors
917-291-5744
- Jim Geraghty joins as Independent Director and Chair, bringing US and international experience from Genzyme and as NED and chair of multiple listed biotechs
- British Patient Capital invests £10 million in a Series B extension bringing total raised in this round to £85.5 million
Oxford, United Kingdom – 15 May 2023 – OMass Therapeutics (‘OMass’, or ‘the Company’), a biotechnology company that identifies medicines against highly validated target ecosystems, today announces the appointment of Jim Geraghty as chairman of its board of directors and an additional investment of £10 million from new investor British Patient Capital (BPC) in a Series B extension, bringing the total raised in this round to £85.5 million.
BPC, a wholly owned commercial subsidiary of British Business Bank plc, the UK government’s economic development bank joins Syncona, Oxford Science Enterprises, GV, Northpond Ventures and Sanofi Ventures in the round. The additional £10 million investment is from BPC’s Future Fund: Breakthrough programme, a £375m programme which co-invests with private sector investors in innovative, R&D-intensive UK companies. It will be used to support OMass as it continues to progress its pipeline of small molecule therapeutics for rare diseases and immunological conditions targeting solute carriers, complex-bound proteins and GPCRs.
Alongside its recent move to new purpose-built 16,000ft2 flagship site on ARC Oxford campus enabling the co-location of its 55-strong and growing workforce, the additional funds will further support OMass as it becomes a clinical stage company, in line with its strategy outlined at the time of its Series B announcement last year.
Catherine Lewis La Torre, CEO, British Patient Capital said: “The UK continues to demonstrate its strength in life sciences with university spinouts like OMass leading the way. Scaling next-generation technology businesses, like OMass, by providing long-term capital for investment is why we established Future Fund: Breakthrough in 2021. We are delighted to be part of this latest funding round which will allow OMass to continue to build a strong pipeline of drugs with the potential to meaningfully improve patient outcomes.”
James (Jim) Geraghty also joins the board of directors to take on the role of independent Chair from Edward Hodgkin Ph.D., who has served as Chairman for nearly five years. Dr. Hodgkin will remain on the board as a Non-Executive Director representing major investor Syncona.
Boston-based Mr. Geraghty is an industry leader with over 35 years of strategic experience including more than 25 years as a senior executive at biotechnology companies developing and commercializing innovative therapies. He is currently chairman on the boards of Orchard Therapeutics (NASDAQ: ORTX) and Pieris Pharmaceuticals (NASDAQ: PIRS) and is a member of the boards of Voyager Therapeutics (NASDAQ: VYGR), Fulcrum Therapeutics (NASDAQ: FULC) and CANbridge Pharmaceuticals (HKEX:1228). Jim served earlier as an entrepreneur-in-residence at Third Rock Ventures, a leading biotech venture fund, and as Senior Vice President Strategy and Business Development at Sanofi. Previously, he served as senior vice president international development at Genzyme, president of Genzyme Europe, and founding president and CEO of Genzyme Transgenics. Mr. Geraghty started his career in healthcare strategy consulting at Bain. A graduate of the Yale Law School, he is also the author of the Nature-reviewed book Inside the Orphan Drug Revolution: The Promise of Patient-Centered Biotechnology.
Jim Geraghty, Chairman of OMass Therapeutics said: “OMass is on a growth trajectory, and I am thrilled to join the board at this exciting time in its journey. OMass’ native mass spectrometry-based platform has great potential, and founder Carol Robinson and CEO Ros Deegan have done great work developing highly promising programs against validated but difficult to drug targets. I look forward to working together with the team and the rest of the board as the company transitions to a development stage company in the near future.”
Ros Deegan, CEO of OMass added: “I am delighted to welcome Jim to our board – his experience in helping to build Genzyme and subsequently with pharma, venture and biotech boards will be invaluable to OMass as we grow, and I look forward to his strategic counsel. I am also pleased to welcome the additional investment and support of British Patient Capital and the confidence this shows in our ambitious growth plans.”

Caption: Jim Geraghty, Chairman of OMass Therapeutics
-ENDS-
For further information, please contact:
|
OMass Therapeutics |
Consilium Strategic Communications |
|
Rosamond Deegan, Chief Executive Officer Phone: +44 (0) 1235 527589 Email: ros.deegan@omass.com |
Sue Charles/Stella Lempidaki/Kumail Waljee Phone: +44 (0)20 3709 5700 Email: omass@consilium-comms.com |
About OMass Therapeutics
OMass Therapeutics is a biotechnology company discovering medicines against highly- validated target ecosystems, such as membrane proteins or intracellular complexes. The company’s unique OdyssION™ technology platform comprises novel biochemistry techniques, next-generation native mass spectrometry, and custom chemistry. This allows OMass to interrogate not just the target, but also the interaction of the target with its native ecosystem, separate from the confounding complexity of the cell. The result is cell-system fidelity with cell-free precision. OMass is advancing a pipeline of small molecule therapeutics in rare diseases and immunological conditions, that target solute carriers, complex-bound proteins, and GPCRs.
Headquartered in Oxford, UK, OMass has raised over $160M (£129M) from a top-tier international investor syndicate, including Syncona, Oxford Science Enterprises, GV, Northpond Ventures, Sanofi Ventures and British Patient Capital.
To learn more, please visit www.omass.com. Follow us on LinkedIn and Twitter.
About British Patient Capital
British Patient Capital Limited is a wholly owned commercial subsidiary of British Business Bank plc, the UK government’s economic development bank. Its mission is to enable long- term investment in innovative firms led by ambitious entrepreneurs who want to build large- scale businesses. Launched in June 2018, British Patient Capital has more than £3bn of assets under management, investing in venture and venture growth capital to support high growth potential innovative UK businesses in accessing the long-term financing they require to scale up. Find out more here.
British Business Bank plc and its subsidiary entities are not banking institutions and do not operate as such. They are not authorised or regulated by the Prudential Regulation Authority (PRA) or the Financial Conduct Authority (FCA). A complete legal structure chart for British Business Bank plc and its subsidiaries can be found on the British Business Bank plc website.
British Patient Capital makes commitments and invests on its own behalf and on behalf of third-party investors whose investments British Patient Capital manages.
The transaction described above does not constitute or imply any endorsement, warranty or recommendation by the UK government, the British Business Bank plc, its subsidiaries or any other party of OMass Therapeutics or its products or services.
- Certain Cyclerion shareholders and new investors have agreed to invest $81M to launch a new company targeting diseases of mitochondrial dysfunction
- Cyclerion to receive $8M in cash and 10% equity in the new company in exchange for its zagociguat and CY3018 assets
- Definitive agreement signing triggers previously announced $5M equity investment in Cyclerion
CAMBRIDGE, Mass., May 11, 2023 (GLOBE NEWSWIRE) -- Cyclerion Therapeutics, Inc. (Nasdaq: CYCN) announced today that it has signed a definitive agreement with a new private company (“NewCo”) to sell two of its sGC stimulator assets in exchange for cash and equity ownership. Investors in NewCo have agreed to invest $81M to develop zagociguat (previously CY6463) to treat MELAS* and other diseases associated with mitochondrial dysfunction and advance CY3018.
Under the terms of the asset purchase agreement, Cyclerion will receive an $8M cash payment at closing, reimbursement for all expenses related to zagociguat and CY3018 for the period between the signing and closing of the transaction, and 10% equity ownership in NewCo that is subject to anti-dilution protection through $100M in post-money valuation. Cyclerion will also have additional future equity purchase rights in NewCo. Current Cyclerion shareholders including Invus and CEO Peter Hecht are participating in the capitalization of NewCo. They are joined in the NewCo investor syndicate by Venrock, J Wood Capital and Sanofi Ventures. The transaction is subject to approval by Cyclerion shareholders. Each of the current Cyclerion investors who are participating in the NewCo capitalization have agreed to vote their Cyclerion shares in favor of the transaction. Following the closing, NewCo will be solely responsible for all activities and expenses related to developing and commercializing zagociguat and CY3018.
Signing of the definitive agreement triggered the previously announced $5M equity investment by CEO Peter Hecht. This investment will take place on May 19, 2023, and Hecht will receive a mix of common stock and nonvoting convertible preferred stock of Cyclerion at a minimum purchase price of $0.434 per share, subject to adjustment for any reverse stock split or similar event.
“We are pleased to see our zagociguat and CY3018 assets attracting the capital and capabilities they will need to continue their development in mitochondrial and CNS diseases. Over the past 12 months, our board, management and advisors have carried out an exhaustive and thorough process to evaluate all available opportunities to maximize the value of our assets for our shareholders in this exceptionally challenging capital market while advancing potentially life-changing medicines to patients,” said Errol De Souza, Chair of the Board at Cyclerion “With this transaction, we believe these compounds will receive the focus they deserve, and Cyclerion shareholders will be able to benefit from future value creation via Cyclerion’s equity position in the new company without any go-forward operational or financial obligations for these programs."
Stifel is acting as financial advisor to Cyclerion; Hughes Hubbard & Reed LLP is serving as legal counsel to the Board and the Company.
About Cyclerion Therapeutics
Cyclerion Therapeutics is a clinical-stage biopharmaceutical company on a mission to develop treatments for serious diseases. Cyclerion’s portfolio includes novel sGC stimulators that modulate a key node in a fundamental signaling network in both the CNS and the periphery. The multidimensional pharmacology elicited by the stimulation of sGC has the potential to impact a broad range of diseases. Zagociguat is a CNS-penetrant sGC stimulator that has shown rapid improvements across a range of endpoints reflecting multiple domains of disease activity, including mitochondrial disease-associated biomarkers. CY3018 is a CNS-targeted sGC stimulator in preclinical development that preferentially localizes to the brain and has a pharmacology profile that suggests its potential for the treatment of neuropsychiatric diseases and disorders. Praliciguat is a systemic sGC stimulator that is licensed to Akebia and being advanced in rare kidney disease. Olinciguat is a vascular sGC stimulator that the Company intends to out-license for cardiovascular diseases. For more information about Cyclerion, please visit https://www.cyclerion.com/ and follow us on Twitter (@Cyclerion) and LinkedIn (www.linkedin.com/company/cyclerion).
Forward Looking Statement
Certain matters discussed in this press release are “forward-looking statements”. We may, in some cases, use terms such as “predicts,” “believes,” “potential,” “continue,” “estimates,” “anticipates,” “expects,” “plans,” “intends,” “may,” “could,” “might,” “will,” “should”, “positive” or other words that convey uncertainty of future events or outcomes to identify these forward-looking statements. In particular, the Company’s statements regarding the potential of zagociguat and CY3018 for the treatment of mitochondrial and CNS diseases, the potential for any successful development of zagociguat or CY3018, any future value creation to the Company from the sale, all of which depend on the successful development, commercialization and/or sales relating to zagociguat and CY3018 (which cannot be assured and is not in the Company’s control), and other trends and potential future results are examples of such forward-looking statements. The forward-looking statements include risks and uncertainties, including, but not limited to, the risks that the Company may never successfully close the referenced transactions, that if completed, the referenced transactions may not be successful in generating future value for Cyclerion shareholders, that zagociguat and CY3018 may not demonstrate the desired safety and efficacy in ongoing and future clinical trials, the ability of the new company to successfully develop and/or commercialize zagociguat and CY3018, and the receipt of regulatory approvals. The factors discussed herein could cause actual results and developments to be materially different from those expressed in or implied by such statements. The forward-looking statements are made only as of the date of this press release and the Company undertakes no obligation to publicly update such forward-looking statements to reflect subsequent events or circumstance.
* MELAS (Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes syndrome)
Investors and Media Inquiries
Cyclerion Investor Relations
Phone: 857-327-8778
Email: IR@cyclerion.com

- $35 million in upfront financing with the potential to receive additional aggregate financing up to $105 million, subject to achieving clinical development milestones, plus up to $45 million upon exercise of warrants
- Led by BVF Partners LP and Armistice Capital, with participation from new investor Sanofi (via Sanofi Ventures)
- Aggregate financing (subject to milestones) expected to be sufficient to fund Company through the completion of the Phase 2 BESTOW trial evaluating tegoprubart for the prevention of rejection in patients receiving a kidney transplant
IRVINE, Calif., May 1, 2023 (GLOBE NEWSWIRE) – Eledon Pharmaceuticals, Inc. (“Eledon”) (NASDAQ: ELDN), a clinical stage transplant and immunology-focused biopharmaceutical company, today announced that it has entered into a definitive securities purchase agreement with certain healthcare investors that will provide up to $185 million in gross proceeds to Eledon through a private placement. The purchase is comprised of an initial upfront financing of $35 million in exchange for 15.2 million common shares (or pre-funded warrants), representing a purchase price of $2.31 for each share of common stock and associated warrant sold at the initial closing, and up to an additional $105 million in mandatory tranche financing, subject to achieving specified milestones, including clinical development milestones. In addition, Eledon will have the potential to receive $45 million upon the full exercise of warrants being issued in connection with the agreement.
The financing is being led by BVF Partners LP and Armistice Capital, and includes participation from new and existing investors including Sanofi (via Sanofi Ventures).
“This financing represents a significant commitment by our shareholders to advance the development of tegoprubart in kidney transplantation,” said David-Alexandre C. Gros, M.D., Chief Executive Officer. “Eledon is now financially positioned to complete and report data from our planned Phase 2 BESTOW study, as well as to continue accumulating and reporting data from our ongoing Phase 1b kidney transplantation trial. We look forward to progressing the development of tegoprubart as a much- needed potential treatment option to better protect and extend the functional life of transplanted kidneys that patients often wait years to receive.”
In addition to the $35 million in upfront financing, the transaction includes a second and third closing, each having mandatory funding conditions whereby the holders have committed to exercise the warrants subject to the satisfaction of certain clinical trial milestones and volume weighted average
share price levels, and trading volume conditions. The private placement also includes a 5-year term warrant with an exercise price of $3.00 that is exercisable at investors’ election.
SVB Securities is acting as lead placement agent. Cantor, LifeSci Capital and Noble Capital Markets are acting as co-placement agents in connection with the financing.
The Company intends to use the net proceeds from the private placement to fund the clinical development of its lead asset tegoprubart, working capital and general corporate purposes.
The securities sold in the private placement, including the common shares underlying the warrants, are being made in a transaction not involving a public offering and have not been registered under the Securities Act of 1933, as amended, or applicable state securities laws and may not be reoffered or resold in the United States except pursuant to an effective registration statement or an applicable exemption from the registration requirements of the Securities Act and applicable state securities laws. Concurrently with the execution of the securities purchase agreement, Eledon and the investors entered into a registration rights agreement pursuant to which the Company has agreed to file a registration statement with the Securities and Exchange Commission registering the resale of the securities issued in the private placement.
This press release shall not constitute an offer to sell or a solicitation of an offer to buy these securities, nor shall there be any sale of these securities in any state or other jurisdiction in which such offer, solicitation or sale would be unlawful prior to the registration or qualification under the securities laws of any such state or other jurisdiction.
About Eledon Pharmaceuticals and tegoprubart
Eledon Pharmaceuticals is a clinical stage biotechnology company using its immunology expertise in targeting the CD40 Ligand (CD40L, also called CD154) pathway to develop therapies to protect transplanted organs and prevent rejection, and to treat ALS. The company’s lead compound in development is tegoprubart, an anti-CD40L antibody with high affinity for CD40 Ligand, a well-validated biological target with broad therapeutic potential. Eledon is headquartered in Irvine, CA. For more information, please visit the company’s website at www.eledon.com.
Forward-Looking Statements
This press release contains forward-looking statements that involve substantial risks and uncertainties. Any statements about the completion of the private placement, the satisfaction of the mandatory funding conditions, exercise of the warrants, other customary closing conditions related to the private placement, the intended use of net proceeds from the private placement, statements about planned clinical trials, the Company’s expectation that the aggregate financing (subject to milestones) is expected to be sufficient to fund the Company through the completion of the Phase 2 kidney transplant trial, and the Company’s other future expectations, plans and prospects, as well as other statements containing the words “believes,” “anticipates,” “plans,” “expects,” “estimates,” “intends,” “predicts,” “projects,” “targets,” “looks forward,” “could,” “may,” and similar expressions, constitute forward- looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Forward- looking statements are inherently uncertain and are subject to numerous risks and uncertainties, including: risks related to the private placement, risks relating to the safety and efficacy of our drug candidates; risks relating to clinical development timelines, including interactions with regulators and clinical sites, as well as patient enrollment; risks relating to costs of clinical trials and the sufficiency of the company’s capital resources to fund planned clinical trials; and risks associated with the impact of the ongoing coronavirus pandemic. Actual results may differ materially from those indicated by such forward-looking statements as a result of various factors. These risks and uncertainties, as well as other risks and uncertainties that could cause the company’s actual results to differ significantly from the forward-looking statements contained herein, are discussed in our quarterly 10-Q, annual 10-K, and other filings with the U.S. Securities and Exchange Commission, which can be found at www.sec.gov. Any forward-looking statements contained in this press release speak only as of the date hereof and not of any future date, and the company expressly disclaims any intent to update any forward-looking statements, whether as a result of new information, future events or otherwise.
Investor Contact:
Stephen Jasper
Gilmartin Group
(858) 525 2047 stephen@gilmartinir.com
Source: Eledon Pharmaceuticals
- Funding from top-tier syndicate will be used to advance potential first-in-class investigational antibody THN391 into clinical trials.
SOUTH SAN FRANCISCO – April 27, 2023 - Therini Bio, Inc., a biotech company aimed at developing fibrin-targeted therapies to treat inflammatory neurodegenerative and retinal diseases, today announced the close of a $36M Series A financing round. The funding round was co-led by Dementia Discovery Fund, MRL Ventures Fund, the therapeutics-focused corporate venture fund of Merck & Co., Inc., Sanofi Ventures, and SV Health Investors’ Impact Medicine Fund. New investor Eli Lilly and Company participated in the round, with all existing investors including Alzheimer’s Drug Discovery Foundation (ADDF), Dolby Family Ventures, and Foundation for a Better World. The Series A funding brings the total amount raised since inception to $62M.
Therini Bio is developing therapeutic candidates that selectively target the inflammatory component of fibrin, in neurological diseases, including Alzheimer's disease (AD) and multiple sclerosis (MS), as well as in a variety of retinal diseases, such as diabetic macular edema (DME) where destructive inflammation plays a role in the disease process.
The new funding will enable Therini Bio to advance its pipeline of fibrin-targeting therapies and advance its lead antibody THN391, that binds the inflammation-driving component of fibrin that is known to activate immune responses in neurodegenerative and ophthalmologic diseases. Importantly, based on our preclinical studies to date, targeting this region does not impact or diminish fibrin’s critical role in blood clotting and coagulation.
"We are thrilled to announce a top-tier investor syndicate, which will allow us to advance our groundbreaking work in developing fibrin-targeted therapies for diseases driven by chronic inflammation,” said Michael Quigley, Ph.D., President and CEO of Therini Bio. “This funding will enable us to accelerate the development of our lead antibody program targeting inflammatory fibrin in neurodegenerative and retinal diseases. We look forward to advancing our first candidate, THN391, into clinical trials, and expect to announce key safety and proof of mechanism clinical data by the end of 2024."
“We are thrilled to invest in Therini Bio and support their innovative first-in-class approach in developing therapies that selectively target inflammatory fibrin,” said Houman Ashrafian, BM BCh, DPhil, Managing Partner, SV Health Investors. “Therini Bio’s approach in optimizing humanized antibodies to target fibrin inflammatory signals without affecting critical clotting functions is groundbreaking, and we look forward to working with the team to advance development of these potentially novel therapies for patients.”
“Loss of vascular integrity, fibrin deposition and subsequent chronic inflammation are known risk factors in the development and progression of Alzheimer's disease. Therini Bio is targeting the root cause of chronic inflammation by developing a targeted monoclonal antibody that reduces fibrin-mediated inflammation in the vasculature of the brain, inhibiting the activation of microglia, immune cells in the brain,” said Howard Fillit, M.D., Co-Founder and Chief Science Officer at the Alzheimer’s Drug Discovery Foundation (ADDF). “The Company is well-positioned to make a significant impact in the fight against Alzheimer's and other neurodegenerative diseases with this potentially novel biological approach to inflammation."
Additionally, Therini Bio was awarded a $3M non-dilutive funding grant in 2021 from the National Institutes of Health (NIH) National Institute on Aging (NIA), which will provide $1M of preclinical research funding to advance the Company’s Alzheimer’s disease program each year through 2024.
About Therini Bio, Inc.
Therini Bio is a biotech company aimed at developing fibrin-targeted therapies to treat inflammatory neurodegenerative and retinal diseases. The Company is developing a pipeline of potential first-in-class therapies targeting toxic fibrin accumulation. The foundational science was licensed based on technology discovered in Katerina Akassoglou, Ph.D. laboratories at the Gladstone Institutes at the University of California San Francisco (UCSF) and formerly the University of California San Diego (UCSD). For more information, visit www.therinibio.com.
NIH Disclosure
Research reported in this release was supported by the NIA of the NIH under Award Number U01AG073125. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Media Contact
Kimberly Ha
KKH Advisors
917-291-5744
NEW YORK, April 25, 2023 ‒ Aetion®, the global leader in real-world evidence (RWE) technology and analytics, is pleased to announce the culmination of the RCT-DUPLICATE demonstration project, with complete findings published today in The Journal of the American Medical Association (JAMA). This three-year initiative tested whether researchers asking clinical questions in real-world data (RWD) — data from patients’ day-to-day interactions with the healthcare system — would obtain similar results to findings from randomized clinical trials (RCTs). In cases where clinical trial designs aligned with real patient care processes, the RWE studies and RCTs came to largely similar conclusions.
As part of the RCT-DUPLICATE initiative, researchers created and published protocols for observational, real-world analogs to thirty completed and two ongoing RCTs, emulating each trial’s design as closely as possible. They then implemented the studies principally using the Aetion Evidence Platform® (AEP), which analyzes data from the real world to produce transparent, rapid, and scientifically validated answers on the safety, effectiveness, and value of medical treatments.
The results show that in about half of the cases where researchers were able to closely mimic the design of the corresponding RCT using RWD, the RWE study came to a similar conclusion as the analogous RCT. In many cases where RWE and RCTs did not come to a similar conclusion, the RCT design itself did not align with real-world clinical practice, creating a challenge for emulation of the trial using RWD. In these instances, RWE and RCTs may both be reaching meaningful conclusions, but to subtly different research questions.
“The RCT-DUPLICATE initiative further demonstrates that RWE has the potential to augment findings from RCTs and guides us to cases where RWE and RCTs may be expected to reach similar conclusions,” said Nicolle Gatto, Ph.D., M.P.H., Chief Science Officer at Aetion. “This work is important in promoting and understanding the value of RWE for decision-making as we continue to assess RWE’s uses, benefits, and limitations.”
Today’s publication, alongside ongoing research like the Coalition for the Advancement of RWE through Randomized Controlled Trial Emulation (CARE) Initiative and other demonstration projects, adds to a body of evidence that demonstrates how RWE can be relied on to broaden our understanding of the safety, effectiveness, and value of medications — complementing or extending what can be demonstrated by RCTs.
To learn more about how RCT-DUPLICATE advances RWE, visit JAMA.
About Aetion®
Aetion is a healthcare analytics company that delivers real-world evidence for the manufacturers, purchasers, and regulators of medical treatments and technologies. The Aetion Evidence Platform® analyzes data from the real world to produce transparent, rapid, and scientifically validated answers on safety, effectiveness, and value. Founded by Harvard Medical School faculty members with decades of experience in epidemiology and health outcomes research, Aetion informs healthcare’s most critical decisions—what works best, for whom, and when—to guide product development, commercialization, and payment innovation. Learn more at aetion.com and follow us on LinkedIn.
Aetion Media Contact
- The ReMMi-D trial will evaluate the efficacy and safety of investigational prescription digital therapeutics for the prevention of episodic migraine in patients 18 years and older.
- Fully decentralized, randomized clinical trial enrolling approximately 558 participants reflects the company’s commitment to innovating digital therapeutics that address high unmet patient need in serious indications, such as migraine.
- The Click Therapeutics platform enables the development and validation of new digital therapeutics that have significant potential to reduce barriers to care and expand patient access to new, additive treatment types that can improve health outcomes.
NEW YORK, April 25, 2023 -- Click Therapeutics, Inc. ("Click"), a leader in Digital Therapeutics™ as prescription medical treatments, today announced the initiation of the ReMMi-D, Reduction in Monthly Migraine Days study, a fully remote and decentralized pivotal trial designed to investigate the efficacy and safety of prescription digital therapeutics in reducing the number of monthly migraine days for adults diagnosed with episodic migraine.
Migraine is a complex and debilitating condition that affects more than 40 million adults1,2, and is the second leading cause of disability in the United States3. Migraine is characterized by episodes of moderate-to-severe headache and generally associated with nausea and increased sensitivity to light and sound.4 Its burden is impacted by employment status, poverty, education attainment and insurance coverage.4,5 Although there are a variety of pharmacologic and nonpharmacologic acute and preventative treatments available, they are not without notable limitations, and patients often do not achieve full remission in response to pharmacotherapy alone.
Migraine is a complex and debilitating condition that affects more than 40 million adults1,2, and is the second leading cause of disability in the United States3. Migraine is characterized by episodes of moderate-to-severe headache and generally associated with nausea and increased sensitivity to light and sound.4 Its burden is impacted by employment status, poverty, education attainment and insurance coverage.4,5 Although there are a variety of pharmacologic and nonpharmacologic acute and preventative treatments available, they are not without notable limitations, and patients often do not achieve full remission in response to pharmacotherapy alone.
Unlike a traditional clinical trial in which a patient receives treatment at a designated clinical trial site, ReMMi-D is a completely remote study in which patients receive digital therapy through a mobile application while going through their regular daily activities and allows for a more representative patient population for enrollment. This study design is consistent with the real-world potential of prescription digital therapeutics to reduce barriers to care by enabling access to therapy for patients anywhere, irrespective of geographic location or proximity to trained specialists.
"Enrolling the first patient in our ReMMi-D pivotal study marks Click Therapeutics’ continued progress toward developing first-in-class, prescription digital therapeutics for patients living with migraine," said Austin Speier, Chief Strategy Officer of Click Therapeutics. "We are driven by our commitment to using digital technology to enable novel approaches that can provide additional treatment options to patients suffering with migraine, fulfilling unmet patient needs and delivering on better patient outcomes."
The Phase 3, randomized, double-blind trial will enroll approximately 558 patients in the United States. Trial intervention will be delivered over 12-weeks and efficacy will be evaluated between groups as a change from baseline in the number of monthly migraine days (MMDs).
"We are pleased and excited to have launched a pivotal trial to validate new ways of helping patients with migraine," said Shaheen Lakhan, MD, PhD, FAAN, Chief Medical Officer of Click Therapeutics. "We are hoping to support not only the 40 million people with migraine, but also the extensive network of primary care providers and fellow neurologists who are at the forefront battling this disease."
Click will present an abstract titled Positive establishment of a digital working alliance with a prescription digital therapeutic in patients with migraine at the 75th Annual Meeting of the American Academy of Neurology (AAN) taking place in Boston from April 22nd to 27th. The team will be presenting Click’s migraine phase 2, Clinical Learning Study (CLS) results, introducing the concept of PDTs to fellow neurologists, and sharing data on how a positive Digital Working Alliance results in robust engagement.
"Our team has worked diligently to develop this groundbreaking technology and we are excited to be at the forefront of medicine's digital revolution," said Han Chiu, Chief Technology Officer of Click Therapeutics. "We believe that our platform-based approach harnessing the power of data, AI, and exceptional product design will lead to a best-in-class treatment for migraine that can improve the lives of patients worldwide."
FDA Breakthrough Device Designation
In December 2022, Click Therapeutics received Breakthrough Device Designation from the U.S. Food and Drug Administration (FDA) for the adjunctive preventive treatment for episodic migraine in patients aged 18 years and older. The Breakthrough Devices Program is intended for devices that have potential to provide more effective treatment over the existing standard of care for life-threatening or irreversibly debilitating diseases. The program is designed to expedite the development and review of medical devices meeting Breakthrough criteria in the United States.
About Click Therapeutics
Click Therapeutics, Inc., develops, validates, and commercializes software as prescription medical treatments for people with unmet medical needs. As a leading innovator of Digital Therapeutics™, we deliver accessible, clinically proven, FDA-regulated prescription treatments to the smartphone in your hand. Our treatments are defined by our commitment to applying technical and scientific rigor and patient-centric design to the development process. This results in uniquely engaging experiences that achieve compelling clinical outcomes for patients seeking new treatment options. We are continuously expanding and refining our shared platform technologies with novel cognitive, behavioral and neuromodulatory mechanisms of action and advanced data-driven tools such as artificial intelligence and machine learning. Digital therapeutics under development on our platform address diverse areas of therapeutic need, including indications in psychiatry, neurology, oncology, cardiology and immunology. Consistently named a best place to work, we foster an inclusive, diverse workforce of innovators, clinicians, scientists, researchers, designers, technologists, engineers and more, united in our mission to provide patients everywhere access to safe and effective prescription digital therapeutics.
For more information, visit www.clicktherapeutics.com and connect with us on LinkedIn.
References
- Katsarava, Z., Buse, D.C., Manack, A.N. & Lipton, R.B. (2012). Defining the differences between episodic migraine and chronic migraine. Curr Pain Headache Rep, 16(1), 86-92. https://doi.org/10.1007/s11916-011-0233-z
- GlobalData. (2021). Migraine – Epidemiology Forecast to 2030. https://www.globaldata.com/store/report/migraine-epidemiology-analysis/
- American Migraine Foundation. (2020). How to Apply for Social Security Disability Insurance with Migraine https://americanmigrainefoundation.org/resource-library/ssdi-migraine/ [Accessed 09/29/2022]
- Torres-Ferrús, M., Ursitti, F., Alpuente, A., Brunello, F., Chiappino, D., de Vries, T., Di Marco, S., Ferlisi, S., Guerritore, L., Gonzalez-Garcia, N., Gonzalez-Martinez, A., Khutorov, D., Kritsilis, M., Kyrou, A., Makeeva, T., Minguez-Olaondo, A., Pilati, L., Serrien, A., Tsurkalenko, O., Van den Abbeele, D., van Hoogstraten, W.S. & Lampl, C. On behalf of School of Advanced Studies of European Headache Federation (EHF-SAS). (2020). From transformation to chronification of migraine: pathophysiological and clinical aspects. J Headache Pain 21, 42 (2020). https://doi.org/10.1186/s10194-020-01111-8
- Burch R. (2021). Preventive Migraine Treatment. Continuum (Minneapolis, Minn.), 27(3), 613–632. https://doi.org/10.1212/CON.0000000000000957. Erratum in: Continuum (Minneap Minn). 2021 Oct 1;27(5):1494-1495. PMID: 34048395
View source version on businesswire.com: https://www.businesswire.com/news/home/20230425005726/en/
Contacts
Investor
Daniel Busby
Media
Jonni Mills
- Purpose-Built Actigraphy Solution Offers a New Standard for Reliable and Convenient Analysis of Sleep and Activity through Empatica's Proven Health Monitoring Platform
BOSTON, April 24, 2023 -- Empatica, a digital health and AI company developing medical-grade wearables and digital biomarkers for health monitoring and diagnostics, today announced the launch of Empatica Actigraphy, a powerful new addition to the company's FDA-cleared Empatica Health Monitoring Platform, offering a new set of digital biomarkers for monitoring sleep and physical activity along with the capability to collect data continuously for up to 14 days on a single charge. The new offering premiered at the American Academy of Neurology's 2023 Annual Meeting, the world's leading gathering of neurology leaders.

Empatica expands its Health Monitoring Platform with a new suite of sleep and activity digital biomarkers
"We believe our community of leading researchers and health care professionals deserve the most reliable, accurate and powerful tools for collecting clinical data" said Matteo Lai, co-founder and CEO of Empatica. "Today, they can access more than twenty new digital measures focused on sleep and activity monitoring on our existing FDA-cleared platform and patient-centric technology. We are proud to continue our work to set the standard for digital health measurements."
Empatica Actigraphy offers two distinct "sensor modes" for EmbracePlus, Empatica's advanced smartwatch for continuous health monitoring: Actigraphy Pro and Actigraphy Optimized. These hardware configurations are designed to tackle commonly faced challenges in clinical research and lower the burden of the patient experience, to improve compliance and participation while generating meaningful data over extended time periods.
Empatica Actigraphy was specifically developed to suit the needs of sleep and activity-related studies and provide digital biomarkers across various core areas including:
- Sleep analysis
- Sleep detection
- Gait/step count
- Wearing detection
- Body position
- Activity intensity
- Energy expenditure
- Activity counts
- Activity classification
Empatica's Platform adds key sleep and activity measures to an already rich offering of digital biomarkers including pulse rate, SpO2 and respiratory rate, making it a highly scalable solution beyond the capabilities of other actigraphy options currently available in the market.
Empatica Actigraphy is available today for use in clinical trials. To learn more about utilizing Empatica Actigraphy and the Empatica Health Monitoring Platform, please visit us at https://www.empatica.com/embraceplus/actigraphy/
About Empatica
Empatica Inc is a pioneer in continuous, unobtrusive remote health monitoring driven by AI. Empatica's platform and technology are used by thousands of institutional partners for research purposes, in studies examining stress, sleep, epilepsy, migraine, depression, addiction, and other conditions. Its flagship medical wearable, EmbracePlus, has been developed with key partners including HHS, USAMRDC, and the NASA-funded TRISH.
Contact:
Steven Burk, sburk@realchemistry.com, +1 (650) 387-2774
- Move to new purpose-built 16,000ft2 flagship site on ARC Oxford campus to support continued company growth
- Expansion of senior leadership team with the appointment of Winfried Barchet Ph.D. as Vice President of Immunology
Oxford, United Kingdom – 19 April 2023 – OMass Therapeutics (‘OMass’, or ‘the Company’), a biotechnology company that identifies medicines against highly validated target ecosystems, today announces the next phase of its growth towards becoming a fully integrated biopharmaceutical company. This includes a move to purpose-designed R&D and office facilities, and expansion of its senior leadership team with the appointment of Winfried Barchet Ph.D. as its Vice President of Immunology.
OMass has expanded over the past 12 months, with a total headcount of 55 employees. The new flagship site offers 16,000ft2 of mixed-use space and will enable co-location and enhanced collaboration of discovery research teams from OMass’ former sites in Oxford and Nottingham. The laboratory space has been refurbished by ARC to meet OMass’ scientific and technical requirements, including air handling for mass spectrometry and cell culture, and new facilities for chemistry and structural biology to support ongoing and future drug discovery efforts. Office space has also been designed to better enable collaborative working both on-site and virtually.
David Williams, Director of Leasing at ARC said: “We’re delighted to be supporting OMass Therapeutics on its growth journey. It’s a leading UK biopharma company at the forefront of innovation in drug discovery. The new building keeps it at the heart of a vibrant ecosystem of innovative companies in Oxford and offers access to communal, social and co-working spaces and a curated programme of events for campus members.”
The Company also continues to expand its senior leadership team and enhance depth of immunology expertise with the recent appointment of Winfried Barchet Ph.D. as its Vice President of Immunology. Winfried brings more than 15 years of project and team leadership experience spanning discovery, translational research and drug discovery in both academia and industry. He joins from IFM Therapeutics where he was the Executive Director of Innate Immunity focusing on drugging targets in the inflammasome and the nucleic acid sensing pathways. Prior to that, he worked at Roche as a discovery area leader in innate immunity and in academia at the University of Bonn where he was the Professor of Translational Immunology at the German Centre for Infection Research.
Ros Deegan, CEO of OMass said: “It is an exciting period of growth for OMass Therapeutics as we move to our new premises, bringing all our staff under one roof, and welcome Winfried to the senior leadership team. Winfried brings a wealth of expertise in immunology as we continue to execute on our plans to discover drugs for previously undruggable targets, and ultimately, bring life changing medicines to patients.”
For further information, please contact:
OMass Therapeutics
Rosamond Deegan, Chief Executive Officer
Phone: +44 (0) 1235 527589
Email: ros.deegan@omass.com
Consilium Strategic Communications
Sue Charles/Stella Lempidaki/Kumail Waljee
Phone: +44 (0)20 3709 5700
Email: omass@consilium-comms.com
About OMass Therapeutics
OMass Therapeutics is a biotechnology company discovering medicines against highly-validated target ecosystems, such as membrane proteins or intracellular complexes. The company’s unique OdyssION™ technology platform comprises novel biochemistry techniques, next-generation native mass spectrometry, and custom chemistry. This allows OMass to interrogate not just the target, but also the interaction of the target with its native ecosystem, separate from the confounding complexity of the cell. The result is cell-system fidelity with cell-free precision. OMass is advancing a pipeline of small molecule therapeutics in rare diseases and immunological conditions, that target solute carriers, complex-bound proteins, and GPCRs.
Headquartered in Oxford, UK, OMass has raised over $150M (£119M) from a top-tier international investor syndicate, including Syncona, Oxford Science Enterprises, GV, Northpond Ventures, and Sanofi Ventures.
To learn more, please visit www.omass.com. Follow us on LinkedIn and Twitter.
About ARC Oxford Campus
Owned by Brookfield, ARC Oxford is part of ARC’s science and tech network of clusters, that includes Harwell Science and Innovation Campus, ARC West London and ARC Uxbridge. The company has five million sq. ft of development potential planned across the UK’s Golden Triangle, with three million sq. ft planned at the 700-acre Harwell site.
To learn more, please visit https://www.arcgroup.io/oxford/. Follow us on LinkedIn and Twitter.
Related Links
- Expands development of novel GBS vaccine in older adult population
- Targeting significant and growing unmet medical need of GBS infection
Copenhagen, Denmark, 17 April 2023 – MinervaX ApS, a privately held Danish biotechnology company developing a novel vaccine against Group B Streptococcus (GBS), announces the first phase 1 clinical study in older adults of its novel GBS vaccine, at CEVAC (Centre for Vaccinology) in Ghent, Belgium.
GBS is a global unmet medical burden and can cause serious illness in people of all ages, worldwide. It is normally associated with infection in pregnant women and new-born babies; however, invasive GBS disease in adults has been increasing over the last 40 years. The older adult population (>65 years of age) and adults with underlying chronic health conditions (diabetes mellitus, cancer, immune suppression, obesity) are at particular risk of invasive GBS disease. There is currently no vaccine available.
The clinical study will investigate the safety and immunogenicity of two dose levels on the MinervaX novel GBS vaccine in an older adult population from 55 to 75 years of age, with and without underlying medical conditions. The trial will investigate the safety and immune response to the dose level currently under development for use in pregnant women (50 μg of each fusion protein) and a higher dose of 125 μg of each fusion protein. Since older adults, certainly those with comorbidities, often mount a less strong immune response than a younger population, up to three doses will be investigated in this trial [ clinicaltrials.gov under the identifier NCT05782179].
MinervaX has completed enrolment and dosing of its 2nd phase II clinical trial of its novel GBS vaccine in pregnant women across Denmark, the UK and South Africa. Details of MinervaX’s ongoing clinical trials can be found at clinicaltrials.gov under the identifiers NCT04596878 and NCT05154578.
Lidia Oostvogels, Chief Medical Officer of MinervaX, said: “Expanding the development of our GBS vaccine for use in an older adult population, including people with increased risk for GBS due to underlying co- morbidities, is a very important step for MinervaX in the battle against this pathogen. This builds on our efforts and experience to develop a product to provide protection to the most vulnerable populations, i.e., neonates in our maternal immunization program, and now older adults including those with certain co- morbid conditions.”
Prof. Isabel Leroux-Roels, Principal Investigator at CEVAC, commented: “GBS is known to cause potentially life-threatening infections in Older Adults and currently there is no vaccine available to prevent this. All the team at CEVAC are very happy to contribute to the development of this vaccine for this high-risk population.”
Prof. Paul Heath, Director of the St George’s Vaccine Institute, London and Lead Investigator of MinervaX’s Phase IIb study across Denmark, the UK and South Africa, remarked: “Streptococcus agalactiae is a common commensal in humans and approximately 25% of all adults will be colonised with GBS in the gastrointestinal or genitourinary tracts at any given time. We are aware of the considerable global burden of this invasive GBS disease in babies and pregnant women and of the urgent need for a vaccine to prevent this. More recently, we have become aware of the burden of GBS in non-pregnant adults, particularly in older adults, and those with underlying health conditions such as diabetes mellitus. There is no current mechanism for preventing GBS disease in this growing population, and there is a well recognised morbidity and mortality. The need for a vaccine for this group of people is therefore urgent and the commencement of GBS vaccine trials in this population is therefore an important and welcome development.”
For further information please contact:
MinervaX
Per Fischer | Chief Executive Officer Email: pbf@minervax.com
Optimum Strategic Communications
Mary Clark / Jonathan Edwards/ Zoe Bolt Email: minervax@optimumcomms.com Tel: +44 (0) 203 882 9621
Notes to Editors:
About MinervaX
MinervaX is a Danish biotechnology company, established in 2010 to develop a prophylactic vaccine against Group B Streptococcus (GBS), based on research from Lund University. MinervaX is developing a GBS vaccine for maternal immunization, and now also for vaccination of older adults, likely to have superior characteristics compared with other GBS vaccine candidates in development. The latter are based on traditional capsular polysaccharide (CPS) conjugate technology. By contrast, MinervaX’s vaccine is a protein- only vaccine based on fusions of highly immunogenic and protective protein domains from selected surface proteins of GBS (the Alpha-like protein family). Given the broad distribution of proteins contained in the vaccine on GBS strains globally, it is expected that MinervaX’s vaccine will confer protection against virtually 100% of all GBS isolates. www.minervax.com
About Group B Streptococcus (GBS)
Streptococcus agalactiae or Lancefield’s Group B Streptococcus (GBS) is a common commensal in humans, approximately 25% of all adults will be colonised with GBS at any given time. Invasive GBS disease is normally associated with infection in pregnant women and new-born babies; however, invasive GBS disease in adults has been increasing over the last 40 years. The older adult population (>65 years of age) and adults with underlying chronic health conditions (diabetes mellitus, cancer, immune suppression, obesity) are at particular risk of invasive GBS disease.
Group B Streptococcus disease in non-pregnant adults causes secondary and primary bacteraemia, septic arthritis, endocarditis, prosthetic joint infection, and necrotising myositis and fasciitis.
It is apparent that outside of pregnancy and the neonatal period, GBS infection results in high morbidity and mortality rates. There is no preventative treatment, cases are managed with antibiotics when an infection is diagnosed. There is a clear unmet medical need for a preventative vaccine that could provide protection to all adults but particularly to the older adult population or those at risk of infection due to underlying medical or demographic conditions. In addition, the incidence is increasing and will probably continue to increase with an increasing older adult population and an increase in the prevalence of obesity and type 2 diabetes around the world.
- First patient dosed in Canada in Phase 1 ANQUR study – the first-ever clinical trial to evaluate a therapy that rescues STATHMIN-2 expression in ALS patients
- STATHMIN-2 is a well-validated protein important for neural repair and axonal stability, the expression of which is significantly decreased in nearly all ALS patients
CAMBRIDGE, Mass., April 6, 2023 – QurAlis Corporation, a clinical-stage biotechnology company developing breakthrough precision medicines for amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases with genetically validated targets, today announced that the first patient has been dosed in its Phase 1 clinical trial of QRL-201 for the treatment of ALS (ANQUR). QRL-201 is a first-in-class therapeutic product candidate aiming to restore STATHMIN-2 (STMN2) expression in ALS patients. ANQUR is the first-ever study to evaluate a therapy that rescues STMN2 expression in ALS patients.
“STATHMIN-2 is a well-validated protein important for neural repair and axonal stability and is the most significantly regulated gene by TDP-43 exclusively in humans. Its expression is significantly decreased in nearly all ALS patients and it is the most consistently decreased gene over all sporadic ALS patient data sets. QRL-201 rescues STMN2 loss of function in QurAlis ALS patient-derived motor neuron disease models in the presence of TDP-43 pathology,” said Angela Genge, M.D., FRCP(C), chief medical officer of QurAlis. “QRL-201 recently entered the clinic in Canada and we are pleased to dose our first patient. We look forward to advancing the ANQUR clinical trial of QRL-201 for the treatment of ALS so that we can make a meaningful difference in patients’ lives.”
The first participant in the ANQUR study was dosed at University of Montréal Hospital Centre (CHUM)by Geneviève Matte, M.D.C.M., FRCP(C), assistant clinical professor, Department of Neurosciences, University of Montréal; ALS clinic director, CHUM; principal investigator, University of Montréal Hospital Research Centre (CRCHUM); and an ANQUR study investigator.
“ALS is a serious neurodegenerative disease with limited treatment options. There is great need for therapies that could slow disease progression and improve outcomes. This study has the potential to show QRL-201 could be such a therapy that could potentially benefit ALS patients who have a loss of STMN2 due to TDP-43 pathology,” said Merit Cudkowicz, M.D., M.Sc., director of the Sean M. Healey & AMG Center for ALS, chief of neurology at Massachusetts General Hospital, director and the Julieanne Dorn Professor of Neurology at Harvard Medical School, and member of QurAlis’ Clinical Advisory Board.
ANQUR (NCT05633459) is a first-in-human global, multi-center, randomized, double-blind, placebo-controlled multiple-ascending dose Phase 1 clinical trial designed to evaluate the safety, tolerability, and pharmacokinetics of QRL-201 versus placebo in patients with ALS. The primary objective of the study is to determine the safety and tolerability of multiple doses of QRL-201 in people living with ALS. The ANQUR clinical trial is expected to include 64 study participants with ALS across sites in Canada, the U.S., the United Kingdom, Belgium, the Netherlands, Italy, Germany, and Ireland.
Visit www.clinicaltrials.gov for more information about the ANQUR study.
About STATHMIN-2 and TDP-43
STATHMIN-2 (STMN2) is a well-validated protein important for neural repair and axonal stability, the expression of which is significantly decreased in nearly all ALS patients. Also known as SCG-10, STMN2 is a protein essential for the stabilization of microtubules which form an important component of the cytoskeleton of cells and axons. STATHMIN-2 is highly expressed in human motor neurons, the cells that primarily degenerate in patients suffering from ALS. In animal models, STMN2 deletion was found to cause axonal degeneration and loss of muscle innervation, which is the primary functional deficit that leads to paralysis in ALS patients.
Using human neuronal stem cell models from ALS patients, QurAlis co-founder and former Harvard professor Kevin Eggan, Ph.D., discovered in 2019 that the expression of STMN2 is regulated by TDP-43. The Eggan Lab showed that loss of normal TDP-43 function leads to a highly significant decrease in expression of STMN2 and an impairment in neuronal repair which could be rescued by restoring STMN2 levels. These results were published in Nature Neuroscience.
In addition to nearly all ALS patients, TDP-43 pathology is also associated with approximately 50 percent of patients with frontotemporal degeneration (FTD), the second most common form of dementia; about a third of Alzheimer’s Disease patients; and up to seven percent of Parkinson’s disease patients.
There are currently no cures for ALS or FTD.Limited therapeutic options are available for ALS and FTD patients who are in desperate need for effective therapies.
About QurAlis Corporation
QurAlis is trailblazing the path to conquering amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases with genetically validated targets with next-generation precision medicines. QurAlis’ proprietary platforms and unique biomarkers enable the design and development of drugs that act directly on disease-causing genetic alterations. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a deep pipeline of antisense oligonucleotides and small molecule programs including addressing sub-forms of ALS that account for the majority of ALS patients. For more information, please visit www.quralis.com or follow us on Twitter @QurAlisCo.
Media contact:
Kathy Vincent
Cambridge, MA – March 21, 2023 – NextPoint Therapeutics, a biotechnology company developing a new world of precision immuno-oncology, announced today that Axel Hoos, MD, PhD, has joined the NextPoint Board of Directors. Dr. Hoos currently serves as the Chief Executive Officer of Scorpion Therapeutics and brings a wealth of business, entrepreneurial and biopharmaceutical research and development expertise to the role.
“Dr. Hoos is a recognized pioneer in immuno-oncology and has shaped the checkpoint therapeutic landscape through his work on a new development paradigm for IO medicines and the development of the anti-CTLA-4 antibody ipilimumab, the first FDA-approved checkpoint immunotherapy,” commented Detlev Biniszkiewicz, PhD, CEO of NextPoint Therapeutics. “Our team will benefit greatly from Dr. Hoos’ expertise and perspective as we advance our novel immuno-oncology programs targeting the HHLA2 pathway to the clinic.”
“Immuno-oncology has become a critical component in our treatment armamentarium for cancer patients, and scientists are seeking the next breakthrough medicine to further expand its reach. I am looking forward to working with the executive team and fellow Board members to help NextPoint realize its promise to deliver the next IO breakthrough,” said Dr. Hoos. “NextPoint's work on the HHLA2 pathway has the potential to expand the reach of precision immuno-oncology to a range of patients who do not benefit from currently available therapies.”
Prior to becoming CEO of Scorpion, Dr. Hoos served as Senior Vice President, R&D Governance Chair, and Therapeutic Area Head for Oncology at GlaxoSmithKline Pharmaceuticals (GSK). In that role, Dr. Hoos built the Oncology business across all therapeutic modalities in the focus areas of immuno- oncology, synthetic lethality, tumor cell targeting, epigenetics, and cell & gene therapy. He is a member of the Board of Trustees at the Sabin Vaccine Institute, Director on the Board of TCR2, and member of the Scientific Advisory Board and Co-Director of the Cancer Immunotherapy Consortium at the Cancer Research Institute. Dr. Hoos received his MD in Medicine from Heidelberg University, and his PhD in Molecular Oncology from the German Cancer Research Center. He trained in general surgery at the Technical University of Munich and as a fellow in cancer research at Memorial Sloan-Kettering Cancer Center.
About NextPoint Therapeutics
NextPoint is advancing the field of immuno-oncology through its leading scientific work on the novel HHLA2 pathway. Our innovative approach integrates foundational science with a defined clinical biomarker to deliver a new class of monotherapies for patients who will not benefit from PD-1/L1 inhibitors. Our team of proven drug developers is working closely with our renowned scientific founders
to launch a new world of precision immuno-oncology. To learn more, visit nextpointtx.com.
Contacts
Media Contact
Chelsea Rule
MacDougall Advisors
CRule@macdougall.bio
- EQT Life Sciences, Droia Ventures, and Sanofi Ventures led the round
- Financing supports clinical activities for Company’s two lead programs in ALS and pipeline development
CAMBRIDGE, Mass., March 9, 2023 – QurAlis Corporation, a clinical-stage biotechnology company developing breakthrough precision medicines for amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases with genetically validated targets, today announced it has closed an oversubscribed $88 million Series B financing, bringing the total funds raised to $143.5 million. The financing was led by EQT Life Sciences, investing from the LSP Dementia Fund, Sanofi Ventures, and Droia Ventures, with participation from the ALS Investment Fund and existing investors LS Polaris Innovation Fund, Mission BioCapital, INKEF Capital, Dementia Discovery Fund, Amgen Ventures, MP Healthcare Venture Management, Mitsui Global Investment, Dolby Family Ventures, Mission Bay Capital, and Sanford Biosciences.
The proceeds from the financing will fund clinical development of QRL-201 and QRL-101, the Company’s lead product candidates in ALS. In addition, the financing will support ongoing and planned research, as well as the advancement of QurAlis’ pipeline with therapeutic candidates that target specific components of ALS and genetically related frontotemporal dementia (FTD) pathology and defined ALS patient populations based on both disease-causing genetic mutation(s) and clinical biomarkers. As part of the Series B financing, Cillian King, Ph.D., managing director at EQT Life Sciences, and Laia Crespo, Ph.D., partner at Sanofi Ventures, will join QurAlis’ board of directors.
“This financing reflects significant investor confidence in the science behind QurAlis’ next-generation precision medicines, world-class team, and commitment to bringing new therapies to patients suffering from ALS and other neurodegenerative diseases,” said Anne C. Whitaker, chair of QurAlis’ board of directors.
“We are fortunate to be funded by this outstanding group of investors who share our commitment to patients with neurodegenerative diseases and our vision to halt disease progression and significantly improve outcomes,” said Kasper Roet, Ph.D., CEO and co-founder of QurAlis. “This financing round recognizes our scientific track record and will help us advance the clinical development of our two lead programs in ALS and robust pipeline through near-term value-creating milestones. We are breaking through the barriers of science in our quest to bring much-needed precision therapies to patients.”
“QurAlis stands out as a leader in the field of neurodegenerative diseases with its next-generation precision medicines and genetically validated targets,” said Philip Scheltens, M.D., Ph.D., head, EQT Life Sciences’ LSP Dementia Fund. “We are extremely excited to join this distinguished group of investors supporting this world-class team to advance what we believe could become life-changing treatments for patients and their families.”
QRL-201 is a first-in-class therapeutic product candidate aiming to restore STMN2 expression in ALS patients. STMN2 is a well-validated protein important for neural repair and axonal stability, the expression of which is significantly decreased in nearly all ALS patients. QRL-201 rescues STMN2 loss of function in QurAlis ALS patient-derived motor neuron disease models in the presence of TDP-43 pathology. QRL-201 recently entered the clinic in the first-ever clinical trial to evaluate a therapy that rescues STMN2 in people with ALS (ANQUR; NCT05633459).
QRL-201 is the second program in QurAlis’ pipeline to enter the clinic recently. In December 2022, QurAlis announced the Company initiated dosing of QRL-101 in a first-in-human Phase 1 clinical trial (NCT05667779). QRL-101 is a first-in-class selective Kv7.2/7.3 ion channel opener for the treatment of hyperexcitability-induced disease progression in ALS.
About QurAlis Corporation
QurAlis is trailblazing the path to conquering amyotrophic lateral sclerosis (ALS) and other neurodegenerative diseases with genetically validated targets with next-generation precision medicines. QurAlis’ proprietary platforms and unique biomarkers enable the design and development of drugs that act directly on disease-causing genetic alterations. Founded by an internationally recognized team of neurodegenerative biologists from Harvard Medical School and Harvard University, QurAlis is advancing a deep pipeline of antisense oligonucleotides and small molecule programs including addressing sub-forms of ALS that account for the majority of ALS patients. For more information, please visit www.quralis.com or follow us on Twitter @QurAlisCo.
Media contact:
Kathy Vincent
310-403-8951
Cambridge, MA – February 21, 2023 – NextPoint Therapeutics, a biotechnology company developing a new world of precision immuno-oncology, announced today the appointment of Leena Gandhi, MD, PhD, as Chief Medical Officer (CMO).
As CMO, Dr. Gandhi will oversee the clinical development of NextPoint’s precision immuno-oncology programs that target the newly discovered HHLA2 pathway to activate anti-tumor immune responses. Dr. Gandhi brings to the NextPoint team nearly 20 years of experience in immuno-oncology and novel drug development from both the academic research setting and the pharmaceutical industry. She has led pivotal studies demonstrating the utility of PD-L1 as a biomarker for efficacy of anti-PD1 agents in lung cancer as well as studies demonstrating the value of combining immunotherapy and chemotherapy in the treatment of non-small cell lung cancer.
“Leena is a proven leader in immuno-oncology and a brilliant physician-scientist, and our team will benefit greatly from her academic and drug development expertise as NextPoint moves forward in its next stage of growth,” said Detlev Biniszkiewicz, PhD, Chief Executive Officer of NextPoint Therapeutics. “We thank Dr. Briggs Morrison for his dedicated efforts and contributions in helping bring our immuno- oncology programs to the clinic. Briggs Morrison, MD, will step down as Interim CMO, but will continue to advise the company on an ongoing basis.”
“I look forward to working with the NextPoint team to accelerate the development of programs targeting the HHLA2 pathway, a novel tumor-suppressive mechanism with the potential to impact many patients,” commented Dr. Gandhi. “NextPoint’s outstanding team of scientists who have characterized the HHLA2 pathway along with experienced drug developers in the management team makes for the ideal chemistry to usher these novel immuno-oncology programs into the clinic.”
Dr. Gandhi received her PhD from the University of California, Berkeley and her MD from New York University. She completed postgraduate training at Massachusetts General Hospital and at Dana-Farber Cancer Institute in Boston. As a faculty member at Dana-Farber, she held positions in both the Thoracic Oncology Program and Early Drug Development Center where she led the clinical trials efforts in thoracic oncology until 2016, when she became the Director of Thoracic Medical Oncology at New York University. In Dr. Gandhi’s next role as Vice President of Immuno-Oncology Development at Eli Lilly, she led the development of novel immuno-oncology agents across cancer types before returning to Dana- Farber in 2020, where she most recently served as the Director of the Center for Cancer Therapeutic Innovation, a cross-malignancy novel therapeutics hub.
About NextPoint Therapeutics
NextPoint is advancing the field of immuno-oncology through its leading scientific work on the novel HHLA2 pathway. Our innovative approach integrates foundational science with a defined clinical biomarker to deliver a new class of monotherapies for patients who will not benefit from PD-1/L1 inhibitors. Our team of proven drug developers is working closely with our renowned scientific founders to launch a new world of precision immuno-oncology. To learn more, visit nextpointtx.com.
Contacts
Media Contact
Lauren Arnold
MacDougall Advisors
1(617)694-5387
larnold@macdougall.bio

Sanofi Ventures, Medical Excellence Capital, and Astellas Venture Management join syndicate with founder RA Capital Management
Funding supports advancing multiple first-in-class degrader pipeline programs for the treatment of autoimmune, neurological, and other diseases driven by pathogenic extracellular proteins
WALTHAM, Mass., February 16, 2023 – Avilar Therapeutics, a biopharmaceutical company focused on extracellular protein degradation, today announced it has raised additional funding with an expanded syndicate of new investors to support advancing a pipeline of first-in-class extracellular protein degraders. The new financing is an upsize to $75 million of the seed round that had initial funding from Avilar’s founding investor, RA Capital Management, and now includes participation by new investors Sanofi Ventures, Medical Excellence Capital (MEC), and Astellas Venture Management (AVM).
“Avilar is pioneering the next frontier in protein degradation and we are excited that our significant progress over the past year in discovering and advancing extracellular protein degraders has attracted this group of leading investors,” said Daniel Grau, CEO and President of Avilar Therapeutics. “This financing accelerates progression of our expanding pipeline and unlocks new applications of our unique and proprietary discovery engine, positioning the company well for our next stage of growth.”
Proceeds from the financing will be used to advance multiple pipeline programs targeting extracellular proteins that are known to be involved in the pathogenesis of autoimmune, neurological, and other diseases yet remain poorly addressed by currently available approaches. In addition, the funding will be used to grow the company and expand the Avilar platform of proprietary technologies, all custom-built and tailored to designing and building extracellular protein degraders.
Concurrent with this financing, Paulina Hill, PhD, Partner at Sanofi Ventures, will join Avilar’s board of directors. Brian Halak, PhD, from MEC will join as board observer and each of Sanofi Ventures and AVM will also have a board observer seat.
“We are very pleased to welcome this seasoned group of investors to the Avilar board of directors,” said Milind Deshpande, PhD, RA Ventures Venture Partner, Avilar Co-Founder, and Chairman of the Avilar Board of Directors. “Their expertise in company building, capital formation, and drug development will be a tremendous resource to Avilar as the innovation leader in the new field of extracellular protein degradation.”
Avilar is building a novel class of protein degraders that target disease-causing extracellular proteins, thereby extending the reach of protein degradation beyond first generation degraders that target intracellular proteins. The company is focused on the discovery and development of ATACs, or ASGPR Targeting Chimeras, which harness a natural process through which endogenous proteins are internalized into hepatocytes via the asialoglycoprotein receptor (ASGPR) and degraded in the endolysosome.
“We founded Avilar with the vision to build an industry leader in extracellular degradation and we are thrilled with the progress the Avilar team has made delivering on this vision,” said Josh Resnick, MD, MBA, Managing Director at RA Capital and co-lead of RA Capital’s early stage company creation efforts. “Avilar’s platform and scientific team were originally incubated at RAVen, the RA Capital incubator, and exemplify RA Capital’s world class and expanding capabilities in new company formation.”
About Avilar Therapeutics
Avilar Therapeutics is a biopharmaceutical company pioneering the discovery and development of extracellular protein degraders, a new frontier in targeted protein degradation. Avilar develops ATACs (ASGPR Targeting Chimeras), a new class of protein degraders that shuttle disease-causing proteins from circulation to the endolysosome where the unwanted proteins are degraded. Avilar has built a proprietary discovery platform that includes novel, high-affinity, small molecule ASGPR ligands and advanced modeling of the biophysics, pharmacokinetics, and pharmacodynamics of ATAC mediated endocytosis and degradation. This platform enables the modular design and synthesis of ATACs extendable across the extracellular proteome to a wide range of proteins involved in the pathogenesis of human diseases. Avilar is leveraging its ATAC platform to create a broad and diverse pipeline of first-in-class extracellular protein degraders. Avilar is based in Waltham, MA. For more information, please visit www.avilar-tx.com and follow us on Twitter @Avilar_Tx and on LinkedIn.
About RA Capital Management
RA Capital Management is a multi-stage investment manager dedicated to evidence-based investing in public and private healthcare and life science companies that are developing drugs, medical devices, and diagnostics. The flexibility of its strategy allows RA Capital to provide seed funding to startups and to lead private, IPO, and follow-on financings for its portfolio companies, allowing management teams to drive value creation from inception through commercialization. For more information, please visit www.racap.com.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi. Sanofi Ventures invests in early-stage biotech and digital health companies with innovative ideas and transformative new products and technologies of strategic interest to Sanofi. Among these areas are oncology, immunology, rare diseases, vaccines, potential cures in other core areas of Sanofi’s business footprint, and digital health solutions. Find out more: www.sanofiventures.com.
About Medical Excellence Capital
Medical Excellence Capital (MEC) is an early-stage life sciences venture fund that uniquely combines a proprietary global network of leading physician scientists with a team of experienced investment professionals, company builders, and operators. For more information, visit www.medexcelcap.com.
About Astellas Venture Management
Astellas Venture Management (AVM) is the wholly-owned venture capital organization within Astellas, dedicated to supporting pre-clinical, cutting-edge science that can bring VALUE to patients. For over 15 years, AVM has provided equity investments to private, early-stage companies developing therapeutic programs and platform technologies, helping them to advance their innovations faster. AVM is a strategic investor, making investments in science that will enhance the current Astellas R&D pipeline or that could catalyze new directions in discovery research. For more information, visit www.astellasventure.com.
Media Contact:
Kathryn Morris
The Yates Network
914-204-6412
kathryn@theyatesnetwork.com
- Fund focuses on early stage investing, company co-creation, leading financing rounds and is committed to continuing its investment reach by prioritizing companies advancing innovation
Paris, January 11, 2023 – Sanofi Ventures has announced an additional multi-year commitment from Sanofi, with an increase in capital to more than $750 million to the evergreen venture fund. In addition to serving as a financial partner to top-tier early-to-mid-stage portfolio companies, the fund supports future efforts for business development and M&A opportunities within Sanofi. The additional capital, confirmed by the executive committee, will also fuel the expansion and investment capacity of the Sanofi Ventures investment team on a global scale.
Paul Hudson
Chief Executive Officer, Sanofi
“Sanofi’s purpose in chasing the miracles of science reaches far beyond our labs. As we continue to build our best-in-class pipeline, we are investing in early stage companies that share our ambition of delivering transformative science and digital innovation. This capital commitment signals Sanofi’s accelerated ambitions in the venture capital community and our continued desire to collaborate with global innovators in the best interests of patients.”
Jason P. Hafler
Managing Director, Sanofi Ventures
“We are grateful for Sanofi’s support over the past decade and appreciate their renewed commitment to early stage innovations that will fuel the next generation of transformative companies aiming to improve the lives of patients.”
Sanofi Ventures invests in top innovators working in areas including immunology and inflammation, rare diseases, oncology, cell and gene therapy, vaccines, and digital health and data science. The team partners across all stages of the private company lifecycle, from Seed to Series B and beyond, leading financings, serving on boards, and taking pride in working alongside portfolio companies to drive long-term value.
In 2022, Sanofi Ventures closed 10 investments in global therapeutic and digital areas of strategic interest to Sanofi. Since its inception, 80% of investments have been in biotherapeutics and 20% have been in digital health companies.
About Sanofi
We are an innovative global healthcare company, driven by one purpose: we chase the miracles of science to improve people’s lives. Our team, across some 100 countries, is dedicated to transforming the practice of medicine by working to turn the impossible into the possible. We provide potentially life-changing treatment options and life-saving vaccine protection to millions of people globally, while putting sustainability and social responsibility at the center of our ambitions.
Sanofi is listed on EURONEXT: SAN and NASDAQ: SNY
Media Relations
Sandrine Guendoul | + 33 6 25 09 14 25 | sandrine.guendoul@sanofi.com
Sally Bain | + 1 617 834 6026 | sally.bain@sanofi.com
Kate Conway | + 1 508 364 4931 | kate.conway@sanofi.com
Sanofi Investor Relations
Eva Schaefer-Jansen | + 33 7 86 80 56 39 | eva.schaefer-jansen@sanofi.com
Arnaud Delépine | + 33 06 73 69 36 93 | arnaud.delepine@sanofi.com
Corentine Driancourt | + 33 06 40 56 92 21 | corentine.driancourt@sanofi.com
Felix Lauscher | + 1 908 612 7239 | felix.lauscher@sanofi.com
Priya Nanduri | + 1 617 764 6418 | priya.nanduri@sanofi.com
Nathalie Pham | + 33 07 85 93 30 17 | nathalie.pham@sanofi.com
Sanofi Forward-Looking Statements
This press release contains forward-looking statements as defined in the Private Securities Litigation Reform Act of 1995, as amended. Forward-looking statements are statements that are not historical facts. These statements include projections and estimates and their underlying assumptions, statements regarding plans, objectives, intentions and expectations with respect to future financial results, events, operations, services, product development and potential, and statements regarding future performance. Forward-looking statements are generally identified by the words “expects”, “anticipates”, “believes”, “intends”, “estimates”, “plans” and similar expressions. Although Sanofi’s management believes that the expectations reflected in such forward-looking statements are reasonable, investors are cautioned that forward-looking information and statements are subject to various risks and uncertainties, many of which are difficult to predict and generally beyond the control of Sanofi, that could cause actual results and developments to differ materially from those expressed in, or implied or projected by, the forward-looking information and statements. These risks and uncertainties include among other things, the uncertainties inherent in research and development, future clinical data and analysis, including post marketing, decisions by regulatory authorities, such as the FDA or the EMA, regarding whether and when to approve any drug, device or biological application that may be filed for any such product candidates as well as their decisions regarding labelling and other matters that could affect the availability or commercial potential of such product candidates, the fact that product candidates if approved may not be commercially successful, the future approval and commercial success of therapeutic alternatives, Sanofi’s ability to benefit from external growth opportunities, to complete related transactions and/or obtain regulatory clearances, risks associated with intellectual property and any related pending or future litigation and the ultimate outcome of such litigation, trends in exchange rates and prevailing interest rates, volatile economic and market conditions, cost containment initiatives and subsequent changes thereto, and the impact that COVID-19 will have on us, our customers, suppliers, vendors, and other business partners, and the financial condition of any one of them, as well as on our employees and on the global economy as a whole. The risks and uncertainties also include the uncertainties discussed or identified in the public filings with the SEC and the AMF made by Sanofi, including those listed under “Risk Factors” and “Cautionary Statement Regarding Forward-Looking Statements” in Sanofi’s annual report on Form 20-F for the year ended December 31, 2021. Other than as required by applicable law, Sanofi does not undertake any obligation to update or revise any forward-looking information or statements.
Programs target the novel HHLA2 pathway, activating anti-tumor immune responses to find and destroy cancer cells
Cambridge, MA / Leverkusen, Germany – January 10, 2023 – NextPoint Therapeutics, a biotechnology company developing a new world of precision immuno-oncology, announced today that it raised $80 million in Series B financing co-led by Leaps by Bayer, the impact investment arm of Bayer AG, and Sanofi Ventures, the strategic venture capital arm for Sanofi. The financing will be used to advance NextPoint’s two lead precision immuno-oncology programs into the clinic, both targeting the newly discovered HHLA2 pathway to activate anti-tumor immune responses.
Additional new investors in the round include Invus, Catalio Capital Management, Sixty Degree Capital and PagodaTree Partners. Existing investors that took part in the financing include MPM Capital Management, Dana-Farber Cancer Institute’s Binney Street Capital and NextPoint founder Gordon Freeman, PhD. As part of the financing, Rakhshita Dhar, Senior Director of Venture Investments Health at Leaps by Bayer, and Paulina Hill, Partner at Sanofi Ventures, will join the NextPoint Board of Directors.
NextPoint’s programs aim to deliver monotherapies for cancer patients without viable treatment options. While immune checkpoint inhibitors targeting PD-1/L1 have revolutionized cancer treatments, many patients do not benefit from these medications and require novel therapeutic strategies. Similar to PD-L1, the tumor antigen HHLA2 is a member of the B7 receptor family1, is highly expressed on certain hard-to-treat cancers2, and drives avoidance of detection from the immune system. Importantly, HHLA2 is independent of PD-L1 and is often most strongly expressed in PD-L1-negative cancers.
NextPoint’s approach re-activates immune cells in tumors that are suppressed by HHLA2-driven immune evasion.
NextPoint originated from the combined expertise of its academic founders, Gordon Freeman, PhD, of the Dana-Farber Cancer Institute, and XingXing Zang, PhD, of Albert Einstein College of Medicine. Drs. Freeman’s and Zang’s independent discovery and characterization of the HHLA2 pathway formed the basis of the NextPoint approach. The company and its founders have shown in preclinical models and with analysis of existing clinical datasets that the HHLA2 pathway is an important tumor-suppressive mechanism in many patients3,4.
“NextPoint is building a deep understanding of the HHLA2 tumor-specific immune-escape mechanism, with the ultimate goal of establishing standalone treatments in cancers with high HHLA2 expression,” said Detlev Biniszkiewicz, PhD, Chief Executive Officer of NextPoint Therapeutics. “The support of our new investors along with the continued commitment of our existing investors and founders emphasizes our momentum and progress in defining precision immuno-oncology for new patient segments.” Juergen Eckhardt, MD, Head of Leaps by Bayer, commented, “Leaps by Bayer was founded to help solve ten of the world’s biggest challenges in health and agriculture, including preventing and curing cancer. We are thrilled to support NextPoint, an exciting addition to our oncology portfolio, as it works to redefine the treatment landscape of immuno-oncology.”
About NextPoint Therapeutics
NextPoint is advancing the field of immuno-oncology through its leading scientific work on the novel HHLA2 pathway. Our innovative approach integrates foundational science with a defined clinical biomarker to deliver a new class of monotherapies for patients who will not benefit from PD-1/L1 inhibitors. Our team of proven drug developers is working closely with our renowned scientific founders to launch a new world of precision immuno-oncology. To learn more, visit nextpointtx.com.
About Bayer
Bayer is a global enterprise with core competencies in the life science fields of health care and nutrition. Its products and services are designed to help people and the planet thrive by supporting efforts to master the major challenges presented by a growing and aging global population. Bayer is committed to driving sustainable development and generating a positive impact with its businesses. At the same time, the Group aims to increase its earning power and create value through innovation and growth. The Bayer brand stands for trust, reliability and quality throughout the world. In fiscal 2021, the Group employed around 100,000 people and had sales of 44.1 billion euros. R&D expenses before special items amounted to 5.3 billion euros. For more information, go to www.bayer.com.
About Leaps by Bayer
Leaps by Bayer, a unit of Bayer AG, leads impact investments into solutions to some of today’s biggest challenges in health and agriculture. The investment portfolio includes more than 50 companies. They are all working on potentially breakthrough technologies to overcome some specific challenges such as, e.g., developing a sustainable protein supply, reducing the environmental impact of agriculture, preventing or curing cancer, and others. For more information, go to leaps.bayer.com.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi. Sanofi Ventures invests in early-stage biotech and digital health companies with innovative ideas and transformative new products and technologies of strategic interest to Sanofi. Among these areas are oncology, immunology, rare diseases, vaccines, potential cures in other core areas of Sanofi’s business footprint, and digital health solutions. Find out more: www.sanofiventures.com
Contacts Media Contact
Lauren Arnold
MacDougall Advisors
1(617)694-5387
larnold@macdougall.bio
Kira Peikoff
Leaps by Bayer
1(973)791-3348
kira.peikoff@bayer.com
- Zhao R, Chinai JM, Buhl S, et al. HHLA2 is a member of the B7 family and inhibits human CD4 and CD8 T cell function. Proc Natl Acad Sci U S A. 2013 Jun 11;110(24):9879-84.
- Janakiram M, Chinai JM, Fineberg S, et al. Expression, clinical significance, and receptor identification of the newest B7 family member HHLA2 protein. Clin Cancer Res. 2015 May 15;21(10):2359-66.
- Wei Y, Ren X, Galbo PM Jr, et al. KIR3DL3-HHLA2 is a human immunosuppressive pathway and a therapeutic target. Sci Immunol. 2021 Jul 9;6(61):9792.
- Bhatt RS, Berjis A, Konge JC, et al. KIR3DL3 Is an Inhibitory Receptor for HHLA2 that Mediates an Alternative Immunoinhibitory Pathway to PD1. Cancer Immunol Res. 2021 Feb;9(2):156-169.
- Newly appointed CMO with more than 25 years’ experience in vaccine development
- Fast Track status granted by US Food and Drug Administration
- Completion of enrolment of its 2nd phase II clinical trial in pregnant women
Copenhagen, Denmark, 5 January 2023 – MinervaX ApS, a privately held Danish biotechnology company developing a novel vaccine against Group B Streptococcus (GBS), today announces the appointment of Lidia Oostvogels as Chief Medical Officer and provides an update on its novel GBS vaccine.
Lidia Oostvogels brings a wealth of experience in vaccine development with more than 25 years’ experience in clinical development. Prior to joining MinervaX, she was Senior Vice President, Area Head, Infectious Diseases and Senior Vice President, Clinical Development, for Prophylactic Vaccines CureVac AG. Oostvogels worked for GSK plc for more than 12 years, where she was Director of Vaccine Discovery and Development, Clinical, focused on Influenza and Zoster. Prior to GSK, she spent nine years Boehringer Ingelheim in a clinical development role. She gained her medical doctor qualification from Ghent University in Belgium. Oostvogels’ experience will be instrumental as MinervaX progresses its GBS vaccine towards phase III clinical development.
MinervaX completed enrolment of its 2nd phase II clinical trial of its novel GBS vaccine in pregnant women across Denmark, the UK and South Africa. The randomized, multicenter trial will evaluate the safety, tolerability and immunogenicity of one and two doses of its GBS vaccine at different dosing regimens in healthy pregnant women. Details of MinervaX’s clinical trials can be found at clinicaltrials.gov under the identifiers NCT04596878, NCT05154578 and NCT05005247.
MinervaX’s GBS vaccine has been granted Fast Track regulatory status by the US Food and Drug Administration. The process is designed to facilitate the development of investigational treatments that demonstrate the potential to address unmet medical needs in serious or life-threatening conditions. Programs with Fast Track designation can benefit from early and frequent communication with the FDA throughout the entire drug development and review process and marketing application.
This follows the European Medicines Agency’s decision to award Priority Medicine (PRIME) status to the vaccine in September, an initiative that optimizes development and evaluation of medicines targeting an unmet medical need.
Per Fischer, Chief Executive Officer of MinervaX, said: “I am delighted to welcome Lidia Oostvogels to the leadership team. Her extensive track record in vaccine development will be invaluable as we continue to make significant progress with our novel GBS vaccine. We are looking forward to an exciting and pivotal year of development milestones in 2023 as we continue to advance our vaccine for the prevention of the large unmet medical need, Group B Streptococcal infections.”
Lidia Oostvogels, Chief Medical Officer of MinervaX, said: “I am hugely excited to join MinervaX and look forward to contributing to the further development of its GBS vaccine. There is a major need for more options to prevent this disease and I am delighted to be working with the experienced team at MinervaX to bring this important vaccine to populations at risk.”
ENDS
MinervaX ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark VAT no. DK32673287
For further information please contact:
MinervaX
Per Fischer | Chief Executive Officer
Email: pbf@minervax.com
Optimum Strategic Communications
Mary Clark / Manel Mateus / Richard Staines / Zoe Bolt
Email: minervax@optimumcomms.com
Tel: +44 (0) 203 882 9621
Notes to Editors:
About MinervaX
MinervaX is a Danish biotechnology company, established in 2010 to develop a prophylactic vaccine against Group B Streptococcus (GBS), based on research from Lund University. MinervaX is developing a GBS vaccine for maternal immunization, likely to have superior characteristics compared with other GBS vaccine candidates in development. The latter are based on traditional capsular polysaccharide (CPS) conjugate technology. By contrast, MinervaX’s vaccine is a protein-only vaccine based on fusions of highly immunogenic and protective protein domains from selected surface proteins of GBS (the Alpha-like protein family). Given the broad distribution of proteins contained in the vaccine on GBS strains globally, it is expected that MinervaX’s vaccine will confer protection against virtually 100% of all GBS isolates. www.minervax.com
About Group B Streptococcus (GBS)
GBS is responsible for nearly 50% of all life-threatening infections in newborns. At any given time, some 15- 25% of women are spontaneously colonized with GBS, and they run the risk of transmitting the bacteria to their child in the womb, during birth and/or during the first months of life. GBS colonization may lead to late abortions, premature delivery, or stillbirth and, in the newborn child, may result in sepsis, pneumonia or meningitis, all of which carry a significant risk of severe morbidity, long- term disability or death.
Currently, the only preventative strategy available involves the use of intravenously delivered prophylactic antibiotics which do not comprehensively prevent GBS infection in utero or protect against late-onset infection in newborns. Not only is this approach expensive and logistically challenging, it fails to cover all, including the most severe cases in the US and Europe, and is rarely available in resource- limited settings.
The development of a GBS vaccine is also endorsed by Group B Strep Support and Group B Strep International, and GBS has been prioritized by a number of public health organizations. Both increased uptake of immunization among pregnant women and greater awareness of the implications of GBS suggest that a safe and effective vaccine targeting GBS would be well suited to address this unmet need.
- Medisafe achieves major milestone with appropriate registrations, anticipating the dynamic needs of the healthcare industry
BOSTON, Jan. 4, 2023 -- Medisafe, a leading digital therapeutics company specializing in medication engagement, is pleased to announce its Software as a Medical Device (SaMD) technology. This announcement comes just months after achieving ISO 13485:2016 certification, meeting quality management system parameters specific to medical devices, as well as ISO 27001:2013 certification, meeting IT security management system standards specific to medication and digital health platforms. International registrations mark Medisafe's continued leadership in the clinical management and patient engagement space.
As the number of software for the management of disease continues to grow, Medisafe's progression represents both the company's commitment to adhering to regulatory standards and exceeding industry expectations, as well as its leadership in the growth of SaMD.
"We're incredibly proud of our accomplishments over the past year to better serve patients, clinicians, as well as pharma partners," said Omri Shor, co-founder and CEO of Medisafe. "This most recent announcement of registering as a SaMD is not only a critical step in our company's growth, it is also imperative as leaders and innovators in digital therapeutics that we anticipate and solve for evolving customer and industry, regulated and non-regulated, needs."
In addition to these milestones, Medisafe is also continuing to focus on the non-regulated side of its business building solutions that exceed the dynamic needs of pharma, HUBs, providers, and patients—including the recent addition of its Salesforce CRM integration and Digital Document Exchange in 2022. Medisafe's end-to-end Care Connector platform supports pharma's vision for a more digitally-enhanced future through an interoperable, personalized, and patient-centric approach to digital patient connectivity that allows organizations to efficiently engage with patients throughout their medication journey. The value that Medisafe's platform and digital drug companion bring to patients is validated by the more than 10 million registrants who use and rely on the technology to manage their medications.
Medisafe will be on site for the 41st Annual J.P. Morgan Healthcare Conference in San Francisco. To learn more about Medisafe's recent advancements and how we are supporting millions of patients manage their medications, stay engaged in their therapy, and create a virtual support system toward living healthier lives, visit www.medisafe.com.
About Medisafe
Medisafe is the leading medication engagement platform that empowers patients to seamlessly manage their treatment journey. By combining advanced technology and behavior science, Medisafe reimagines the treatment journey to guide patients' specific journey needs and drive daily engagement. Its machine learning technology fuels the holistic patient engagement platform to personalize their support needs in a scalable fashion. By integrating existing patient support programs into its platform to extend capabilities, Medisafe is building a seamless future model of patient support and better health. Over 10M registered patients and caregivers rely on Medisafe's platform, delivering double digit results toward improving outcomes. The company manages over two billion medication doses via iOS and Android smartphones and tablets. With an average rating of 4.7 out of 5 stars and more than 400,000 user reviews, Medisafe helps to create more daily engagement than Facebook or Twitter applications. Medisafe is a HIPAA and GDPR compliant solution and Medisafe is ISO 27001:2013 and ISO 13485:2016 certified.
Ingelheim, Germany and New York, New York, 19 December 2022 - Boehringer Ingelheim and Click Therapeutics today announced the launch of an expanded collaboration for the development and commercialization of a second prescription-based digital therapeutic (PDT). The companies will collaborate to develop and commercialize a novel mobile application, which combines multiple clinically validated therapeutic interventions for use alone and in combination with pharmaceutical therapy to help people with schizophrenia achieve positive clinical outcomes. The partnership aims to provide additional treatment options to those living with schizophrenia, where there remains a significant unmet need due to lack of access to psychosocial intervention therapies.
Schizophrenia is one of the 15 leading causes of disability worldwide, with approximately half of all patients exhibiting co-occurring mental and/or behavioral health disorders.1 It is a serious mental health condition that alters a person’s perception of reality and impacts how they think, feel, and behave, and people diagnosed with schizophrenia can remain functionally impaired due to insufficiently treated negative symptoms, including cognitive deficits and limited social functioning.2 Treatment guidelines recommend tailored psychosocial intervention therapies, however, access to these interventions is limited. 2,3 Providing novel, digital therapeutic options has the potential to significantly improve treatment and positively impact health and patient’s quality of life.
The additional prescription digital therapeutic under this expanded collaboration builds on development and clinical successes together with key patient insights gained under the companies’ existing collaboration, which has been in effect since September 2020. The CT-155 program has achieved all development milestones to-date and generated supportive data across clinical learning studies in advance of an upcoming pivotal registration study. The companies are pursuing this new therapy after recognizing that a comprehensive treatment strategy for schizophrenia would benefit from a multi-product approach. Click Therapeutics will receive an upfront payment, funding for research and development activities as well as clinical, regulatory and commercial milestone payments up to a total of USD 460 million, plus tiered royalties.
“We are very excited to expand our collaboration with Click Therapeutics and further tap into their expertise in developing efficacious and engaging digital therapeutics. This brings us an important step closer to our aim of developing holistic mental health solutions for patients in need. It opens new horizons on our journey toward advancing mental healthcare by combining pharmacotherapies with newly emerging digital technologies,” said Nedim Pipic, MD, Corporate Vice President, Therapeutic Area Head CNS, Retinopathies & Emerging Areas, Boehringer Ingelheim.
“Our collaboration with Boehringer Ingelheim offers significant potential to bring new digital treatment options to a currently underserved patient population,” said David Benshoof Klein, CEO, Click Therapeutics. “Our teams share a passion to pursue new, groundbreaking approaches to treatment and a mission to make a difference for patients. We are thrilled to expand our relationship with Boehringer Ingelheim to additional treatments within schizophrenia.”
“With this new project we are addressing symptoms of patients for which no scalable treatment approaches exist today. Seeing the pipeline of PDTs growing reflects our commitment to mental health. We are excited to realize the potential of complementing pharmacological therapy with digital solutions to transform the lives of patients,” added Cornelia Dorner-Ciossek, Ph.D., Digital Health Lead at Boehringer Ingelheim.
“Expanding our successful partnership with Boehringer Ingelheim enables our team to build on the experience and insights gathered with CT-155 and further expand the scope of our digital therapeutics platform,” said Austin Speier, Chief Strategy Officer, Click Therapeutics. “This growing collaboration is a testament to the progress we have achieved so far working together and creates the inspiring opportunity to expand CT-155 into a seamless digital therapy suite that can deliver meaningful outcomes to people living with schizophrenia.”
About Boehringer Ingelheim
Boehringer Ingelheim is working on breakthrough therapies that improve the lives of humans and animals. As a leading research-driven biopharmaceutical company, the company creates value through innovation in areas of high unmet medical need. Founded in 1885 and family-owned ever since, Boehringer Ingelheim takes a long-term perspective. Around 52,000 employees serve more than 130 markets in the three business areas, Human Pharma, Animal Health, and Biopharmaceutical Contract Manufacturing. Learn more at www.boehringer-ingelheim.com.
About Click Therapeutics
Click Therapeutics, Inc. develops and commercializes software as prescription medical treatments for patients with unmet medical needs. Through cognitive and neurobehavioral mechanisms, Click’s Digital Therapeutics™ enable change within individuals, and are designed to be used independently or in conjunction with biomedical treatments. The Clickometrics® adaptive data science platform continuously personalizes user experience to optimize engagement and outcomes. Following a groundbreaking clinical trial, Click’s industry-leading smoking cessation program is available nationwide through a wide variety of payers, providers, and employers. Click’s lead prescription program has entered a pivotal, fully remote, randomized, controlled trial on the Verily platform for the treatment of Major Depressive Disorder (MDD) in up to 360 adults. Click is progressing a broad pipeline of Digital Therapeutics™ across a variety of high-burden therapeutic areas, including MDD, Schizophrenia, Migraine, MS, Chronic Pain, Atopic Dermatitis, Acute Coronary Syndrome (ACS), Obesity, Oncology and more. For more information on Click, visit www.ClickTherapeutics.com.
References
1. National Institute of Mental Health. (May 2018). Schizophrenia Statistics [Website]. Accessed July 20, 2020.
2. National Institute of Mental Health. (May 2020). Schizophrenia [Website]. Accessed July 20, 2020.
3. Kozloff, N., Mulsant, B. H., Stergiopoulos, V., & Voineskos, A. N. (2020). The COVID-19 Global Pandemic: Implications for People With Schizophrenia and Related Disorders. Schizophrenia bulletin, 46(4), 752–757. https://doi.org/10.1093/schbul/sbaa051
Media Contacts
Boehringer Ingelheim
Reinhard Malin (CET time zone)
Innovation Unit/Bio Comms, Corp. Affairs
P: 49 6132 77-90815
reinhard.malin@boehringer-ingelheim.com
Linda Ruckel (ET time zone)
Innovation Unit/Bio Comms, Corp. Affairs
P: 1 203-791-6672
linda.ruckel@boehringer-ingelheim.com
Click Therapeutics
Daniel Busby (Investor Contact)
Jonni Mills (Media Contact)
New York, December 16, 2022 - Click Therapeutics, Inc. (“Click”), a leader in Digital Therapeutics™ as prescription medical treatments, today announced that it has received Breakthrough Device Designation from the U.S. Food and Drug Administration (FDA) for CT-132. Click’s CT-132 prescription digital therapeutic is under development as an adjunctive preventive treatment for episodic migraine in patients aged 18 years and older.
Migraine is a complex and debilitating condition that affects more than 47 million Americans,1 and is the second leading cause of years lived with a disability.2 For those living with migraine, attacks unfold over hours to days and negatively impact many domains including employment, educational attainment, and relationships.3 Despite its prevalence, migraine remains a disorder with high unmet need due to incomplete remission and lack of access to specialty care.
The Breakthrough Devices Program is intended for devices that have potential to provide for more effective treatment over existing standard of care for life-threatening or irreversibly debilitating diseases. The program is designed to expedite the development and review of medical devices meeting Breakthrough criteria in the United States.
“We are thrilled to receive this Breakthrough designation as it will facilitate collaborative discussions with the FDA and help expedite the process of bringing a first-in-class migraine digital therapeutic to patients,” said Austin Speier, Chief Strategy Officer of Click Therapeutics. “This is also powerful recognition of the innovative work led by our in-house science and development teams to create a new approach to treating migraine, one supported by early, promising clinical data.”
“Breakthrough further affirms that our unique approach to unlock undruggable CNS targets has merit through the combination of digital neuroactivation and modulation (DiNaMo) and neurobehavioral interventions,” said Shaheen Lakhan, MD, PhD, FAAN, Chief Medical Officer of Click Therapeutics. “Through this new paradigm, we aim to restore lives ravaged by debilitating brain diseases like migraine.”
Additionally, The CT-132 program is validated by the active support of Click’s Migraine Advisory Board comprising leading thought-leaders in headache research and clinical care chaired by Stewart Tepper, MD, professor of neurology of Geisel School of Medicine at Dartmouth and director of Dartmouth Headache Center.
Click has completed or initiated three clinical studies on CT-132, leveraging and expanding its proprietary Click Neurobehavioral Intervention (CNI) Platform in the process. When complete, the data from these trials will support the product’s FDA regulatory submission.
About Click Therapeutics
Click Therapeutics, Inc. develops and commercializes software as prescription medical treatments for patients with unmet medical needs. Through cognitive and neurobehavioral mechanisms, Click’s Digital Therapeutics™ enable change within individuals, and are designed to be used independently or in conjunction with biomedical treatments. The Clickometrics® adaptive data science platform continuously personalizes user experience to optimize engagement and outcomes. Following a groundbreaking clinical trial, Click’s industry-leading smoking cessation program is available nationwide through a wide variety of payers, providers, and employers. Click’s lead prescription program has entered a pivotal, fully remote, randomized, controlled trial on the Verily platform for the treatment of Major Depressive Disorder (MDD) in up to 360 adults. Click is progressing a broad pipeline of Digital Therapeutics™ across a variety of high-burden therapeutic areas, including MDD, Schizophrenia, Migraine, MS, Chronic Pain, Atopic Dermatitis, Acute Coronary Syndrome (ACS), Obesity, Oncology and more. For more information on Click, visit ClickTherapeutics.com.
References:
- Stovner, L.J. & GBD 2016 Headache Collaborators. (2018). Global, regional, and national burden of migraine and tension-type headache, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. The Lancet Neurology, Volume 17, Issue 11, 954 - 976. https://doi.org/10.1016/S1474-4422(18)30322-3
- James, S. L., Abate, D., Abate, K. H., Abay, S. M., Abbafati, C., Abbasi, N., … Murray, C. J. L. (2018). Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. The Lancet, 392(10159), 1789–1858. https://doi.org/10.1016/s0140-6736(18)32279-7
- Burch, R., Rizzoli, P., & Loder, E. (2018). The Prevalence and Impact of Migraine and Severe Headache in the United States: Figures and Trends From Government Health Studies. Headache: The Journal of Head and Face Pain, 58: 496-505. https://doi.org/10.1111/head.13281
Contacts
Investor
Daniel Busby: dbusby@clicktherapeutics.com
Media
Jonni Mills: jmills@clicktherapeutics.com
- Financing from new investors Trill Impact Ventures and Pureos Bioventures as well as existing investors
- Backing from European Investment Bank
- Planning for Phase 3 clinical development of novel GBS vaccine
Copenhagen, Denmark, 15 December 2022 – MinervaX ApS, a privately held Danish biotechnology company developing a novel vaccine against Group B Streptococcus (GBS), today announces the successful completion of a 72 million EUR financing round. The equity financing of 22 million EUR was co-led by new investors Trill Impact Ventures and Pureos Bioventures and includes existing investors REPAIR Impact Fund (Novo Holdings), Sunstone Life Science Ventures, LF Investment, Wellington Partners, Sanofi Ventures, Adjuvant Capital and Industrifonden. In addition to the equity financing, the European Investment Bank provided a 50 million EUR loan facility.
MinervaX has had an exciting year of clinical development for its novel GBS vaccine. The Company commenced enrolment of pregnant women in its Phase 2 clinical trial in Denmark, the UK, and South Africa and completed the dosing of healthy adult women, previously dosed with its GBS vaccine, in a Phase 1 booster clinical trial in the UK.
The financing will enable MinervaX to advance the late-stage development of its GBS vaccine candidate in preparation for a Phase 3 clinical trial. The vaccine candidate was recently awarded PRIME status by the European Medicines Agency (EMA) due to its potential to prevent life-threatening infections in newborn babies. MinervaX also aims to expand its clinical development team and evaluate the Phase 2 clinical data for efficacy and safety ahead of publication next year.
Per Fischer, Chief Executive Officer of MinervaX, said: “We are delighted to announce this financing, which gives us the firepower to accelerate the development of our novel GBS vaccine. GBS can be life-threatening for newborn babies and there is no approved, universally available vaccine to date. I would like to thank the investors and the MinervaX team who have worked very hard on our vaccine candidate in preparation for the final stage of clinical development.”
In conjunction with the financing, Bita Sehat, Senior Director at Trill Impact Ventures, and Veronica Gambillara, Partner of Pureos Bioventures, will join the Board of Directors.
Veronica Gambillara, Partner at Pureos Bioventures, commented: “We are excited to join the MinervaX team and support the development of this vaccine which could protect against one of the leading causes of neonatal and infant sepsis. We are impressed with the clinical data previously generated and the dedication of the leadership team. This financing will expedite the development of a product that has the potential to drastically improve the options available to address GBS infections.”
Bita Sehat, Senior Investment Director at Trill Impact Ventures, added: “MinervaX’s GBS vaccine holds great promise to address a large unmet medical need globally by preventing serious infections in both newborns and pregnant women. Its successful development will not only save lives, it will also contribute to combatting antimicrobial resistance. We are happy to join forces with a strong shareholder base and an excellent management team to support turning this promise into reality. We view MinervaX as a great example of societal and commercial impact going hand in hand.”
EIB Vice president Christian Thomsen, responsible for Denmark, added: “We are delighted to be supporting MinervaX with its medical research into the unmet medical need. Being one of the biggest financiers of innovation in Europe, this fits our investment strategy; to support highly innovative biotech companies developing breakthrough Life Science products, which have the potential to transform and improve people’s lives.”
Details of MinervaX’s clinical trials can be found at clinicaltrials.gov under the identifiers NCT04596878, NCT05154578 and NCT05005247.
ENDS
For further information please contact:
MinervaX
Per Fischer | Chief Executive Officer
Email: pbf@minervax.com
Optimum Strategic Communications
Mary Clark / Manel Mateus / Richard Staines/ Zoe Bolt
Email: minervax@optimumcomms.com
Tel: +44 (0) 203 882 9621
Notes to Editors:
About MinervaX
MinervaX is a Danish biotechnology company, established in 2010 to develop a prophylactic vaccine against Group B Streptococcus (GBS), based on research from Lund University. MinervaX is developing a GBS vaccine for maternal immunization, likely to have superior characteristics compared with other GBS vaccine candidates in development. The latter are based on traditional capsular polysaccharide (CPS) conjugate technology. By contrast, MinervaX’s vaccine is a protein-only vaccine based on fusions of highly immunogenic and protective protein domains from selected surface proteins of GBS (the Alpha-like protein family). Given the broad distribution of proteins contained in the vaccine on GBS strains globally, it is expected that MinervaX’s vaccine will confer protection against virtually 100% of all GBS isolates. www.minervax.com
About Group B Streptococcus (GBS)
GBS is responsible for nearly 50% of all life-threatening infections in newborns. At any given time, some 15- 25% of women are spontaneously colonized with GBS, and they run the risk of transmitting the bacteria to their child in the womb, during birth and/or during the first months of life. GBS colonization may lead to late abortions, premature delivery, or stillbirth and, in the newborn child, may result in sepsis, pneumonia or meningitis, all of which carry a significant risk of severe morbidity, long- term disability or death.
Currently, the only preventative strategy available involves the use of intravenously delivered prophylactic antibiotics which do not comprehensively prevent GBS infection in utero or protect against late-onset infection in newborns. Not only is this approach expensive and logistically challenging, it fails to cover all, including the most severe cases in the US and Europe, and is rarely available in resource- limited settings.
The development of a GBS vaccine is also endorsed by Group B Strep Support and Group B Strep International, and GBS has been prioritized by a number of public health organizations. Both increased uptake of immunization among pregnant women and greater awareness of the implications of GBS suggest that a safe and effective vaccine targeting GBS would be well suited to address this unmet need.
About Trill Impact
Trill Impact is a pioneering Impact House with EUR 1,2 billion in assets under management across its investment strategies. With a team of more than 35 experienced professionals based in the Nordics and Germany, Trill Impact aims to become a force for positive change and realize its vision of delivering real returns and lasting impact for the benefit of investors, businesses and society at large.
For further information, please visit www.trillimpact.com
About Pureos Bioventures
Pureos Bioventures is venture capital fund advised by Swiss-based Pureos Partners that invests exclusively in private innovative drug development companies, with a special emphasis on the next generation of biological drugs and drug formats. The fund’s portfolio companies are built on scientific excellence to develop therapies across a broad indication spectrum including oncology, immunology, ophthalmology, rare diseases, and neuroscience. Pureos has built a team with in-depth investment, operating and clinical expertise, that strives to impact patients’ lives by advancing innovative treatments for devastating diseases.
For further information, please visit www.pureosbio.com
About the European Investment Bank
The European Investment Bank (EIB) is the bank of the European Union, owned by the EU27 Member States. It is active in some 160 countries and is the world’s largest multilateral lender for climate action projects. The bank is providing long term financing for economically sustainable investments to contribute to the European Union’s political objectives. The EIB Group has set “ensuring a just transition for all” as one of the four overarching objectives in its Climate Bank Roadmap 2025. The EIB’s ambition is to support €1 trillion of climate action and environmental sustainability investments in the decade to 2030 and align all its new operations with the goals and principles of the Paris Agreement.
About InnovFIN-IDFF
The InnovFin Infectious Diseases Finance Facility (IDFF) provides financial products ranging from standard debt to equity-type financing for amounts typically between €7.5 million and €75 million to innovative players active in developing or manufacturing innovative vaccines, medicines, medical and diagnostic devices or novel research to combat infectious diseases. Project costs may include laboratory-validated technologies, which require clinical testing for further development, in addition to complementary pre-clinical research. The product is available directly through the European Investment Bank and will continue to support innovative infectious diseases projects until the end of 2022.
For further information, please visit www.eib.org
- Financing led by New Enterprise Associates (NEA), Abingworth and Forge Life Science Partners
- Proceeds support advancement of EP262 and EP547 to multiple clinical milestones
SAN DIEGO, Calif., November 28, 2022 – Escient Pharmaceuticals, a clinical-stage company advancing novel small molecule therapeutics for the potential treatment of a broad range of neurosensory-inflammatory disorders, today announced the closing of a $120 million Series C financing.
The financing was co-led by NEA, Abingworth, and Forge Life Science Partners with participation from other new investors Avego, PFM Health Sciences, and The Eleven Fund, as well as the company’s existing investors The Column Group, 5AM Ventures, Redmile Group, Cowen Healthcare Investments, Sanofi Ventures, Osage University Partners (OUP), and Altitude LifeScience Ventures.
Proceeds from the financing will be used to advance the company’s pipeline of two first-in-class product candidates in several indications. This includes clinical proof-of-concept studies for EP262 (MRGPRX2 antagonist) in chronic spontaneous urticaria (CSU), chronic inducibleurticaria (CindU) and atopic dermatitis (AD) and for EP547 (MRGPRX4 antagonist) in cholestatic pruritus.
Escient’s initial focus is on a novel class of cell surface receptors called Mas-related G protein-coupled receptors, or MRGPRs. MRGPRs are chemosensory receptors expressed on sensory neurons and immune cells, which reside in close proximity to one another in key barrier tissues such as the skin, airways and gastrointestinal tract. MRGPRs mediate the neuro-immune overactivation that is a hallmark of numerous chronic disorders, including many allergic, inflammatory, auto-immune, pain and pruritic diseases.
“By specifically blocking the activation of MRGPRX2 and MRGPRX4 with small molecule antagonists, we aim to develop effective oral medications with novel mechanisms of action for serious neurosensory-inflammatory diseases without the serious side effects observed withother approaches,” said Joshua Grass, Chief Executive Officer of Escient Pharmaceuticals. “We are thrilled with the support from both existing and new investors, and this significant financing puts us in a great position to advance our pipeline to clinical proof-of-concept in multiple indications.”
ABOUT ESCIENT PHARMACEUTICALS
Escient Pharmaceuticals is a clinical-stage company focused on developing novel therapeutics to address a broad range of neurosensory-inflammatory disorders. The company’s pipeline includes two first-in-class small molecule antagonists targeting MRGPRX2 for the treatment of various mast cell mediated disorders and MRGPRX4 for cholestatic pruritus. Based in San Diego, California, Escient is led by an experienced management and scientific team and funded by top-tier life science investors.
CONTACT:
Aaron Mishel
Chief Financial Offi cer
Escient Pharmaceuticals, Inc.
info@escientpharma.com
The Empatica Health Monitoring Platform can accelerate the development of novel therapeutics and the adoption of digital endpoints in patient care and clinical trials. The FDA clearance includes data collection for the continuous monitoring of SpO2, Electrodermal Activity, Skin Temperature and activity associated with movement during sleep.
Boston, MA — Empatica, a digital health and AI company developing medical-grade wearables and algorithms for health monitoring and diagnostics, today announced the clearance of its Empatica Health Monitoring Platform by the U.S. Food and Drug Administration (FDA).
The Empatica Health Monitoring Platform is a full-stack remote health monitoring and data collection solution for research and healthcare professionals, built on data collected by Empatica’s medical-grade, CE-certified EmbracePlus wearable. In addition, the Platform includes Empatica’s proprietary Care software suite, secure cloud infrastructure, and clinically validated digital biomarkers.
Empatica’s Platform has been cleared for continuous data collection to monitor blood oxygen saturation (SpO2) during rest, peripheral skin temperature, activity associated with movement during sleep, and Electrodermal Activity (EDA). Each of Empatica’s digital biomarkers is based on trained algorithms that analyze sensor data in one-minute intervals, and have been rigorously validated in clinical studies conducted with diverse groups of participants. Platform users can also access raw data collected by EmbracePlus’ five sensors, and research-grade digital biomarkers such as Pulse Rate, Pulse Rate Variability, and Respiratory Rate.
The Empatica Health Monitoring Platform is being used globally by major pharmaceutical companies to continuously gather and analyze physiological data for clinical trials evaluating the impact of novel therapeutics, with Empatica collaborating to develop digital biomarkers for use as endpoints. Researchers also have the possibility to develop their own digital biomarkers, which they can implement in their digital health applications or infrastructure using the Platform’s Software Development Kit.
“This clearance represents a significant step forward for our scientific community,” said Dr. Marisa Cruz, Chief Medical Officer of Empatica. “Patients, healthcare providers, and researchers deserve digital health products that are accurate, validated in diverse populations, and intuitive to use. We are proud to have built a solution that accomplishes these goals, offering a high-quality and reliable digital health tool to scientists working to improve patient outcomes through research and clinical care.”
Today Empatica also announced the recent closing of its Series B financing, led by Sanofi Ventures and RA Capital, and participation by Black Opal Ventures. The investment will enable Empatica to expand its suite of digital biomarkers, for use in patient care and as digital endpoints in clinical trials.
“We are excited to team up with Empatica, their investors and partners on this journey.” said Cris De Luca, Partner at Sanofi Ventures and newly-appointed Empatica board member. “By gaining higher resolution into disease symptomology through novel digital measures and digital biomarkers in clinical and real-world settings, Empatica is unlocking the possibilities of early disease detection, enhanced treatment decisions, and improving quality of life for patients around the world”.
Empatica – www.empatica.com
Empatica Inc is a pioneer in continuous, unobtrusive remote health monitoring driven by AI. Empatica's platform and technology are used by thousands of institutional partners for research purposes, in studies examining stress, sleep, epilepsy, migraine, depression, addiction, and other conditions. Its flagship medical wearable, EmbracePlus, has been developed with key partners including HHS, USAMRDC, and the NASA-funded TRISH.
- Joint publication in Nature Chemistry between OMass Therapeutics’ scientists and co-founder Professor Dame Carol Robinson’s team at Oxford University
- Data demonstrates the ability of native mass spectrometry (MS) to interrogate the pharmacology of the beta-1 adrenergic receptor (β1AR), a G protein-coupled receptor (GPCR)
- Discovery of endogenous zinc ion as a positive allosteric modulator in well studied receptor exemplifies the potential of native MS to uncover novel insights that may be important for drug discovery
Oxford, United Kingdom – 10 November 2022 – OMass Therapeutics (‘OMass’, or ‘the Company’), a biotechnology company that identifies medicines against highly validated target ecosystems, today highlights the potential benefits of native mass spectrometry (MS) in drug discovery through its new publication “Mass spectrometry captures biased signalling and allosteric modulation of the β1 adrenergic receptor” published today in Nature Chemistry¹.
In this publication, the authors demonstrate how native MS can be used to investigate the pharmacology of GPCRs using β1AR as a model system. Being able to combine the sensitivity and near atomic mass resolution of the mass spectrometer, whilst preserving the receptor-G protein interactions, allowed the authors to monitor, with high sensitivity, the full spectrum of receptor pharmacology in vitro. Due to mass differences between different G proteins, the authors were also able to distinguish if a ligand is able to promote biased signalling towards a specific pathway.
The authors also discovered an endogenous zinc molecule associated with the receptor. This cation was shown to be a positive allosteric modulator of β1AR and demonstrates the potential for native MS to uncover novel insights that may be important for drug discovery. As an example, the regulation of zinc binding or mimicking its mechanism with a small molecule may provide a new avenue for modulating the kinetics or signalling of β1AR for therapeutic purposes.
These examples highlight two key benefits of utilizing native MS within drug discovery, namely: reconnecting the two fundamental drivers of pharmacology, binding and function; and interrogation of a native ecosystem with high precision.
Professor Dame Carol Robinson, co-founder of OMass Therapeutics and Chair of the Scientific Advisory Board said: “Our findings demonstrate that native MS can be used to reconnect the two fundamental drivers of pharmacology, binding and function. From a practical viewpoint, GPCRs had represented a major challenge for native MS. Overcoming these difficulties has allowed us to monitor attenuated G-protein coupling, driven by a wide range of ligands, highlighting the sensitivity and robustness of our approach.
I am also delighted that despite the fact that β1AR is a well-studied receptor, we were able to ;find a previously unknown endogenous zinc molecule associated with the receptor, which could have implications for the design of new allosteric modulators.”
Originally spun out of Oxford University, OMass has commercialised Professor Dame Carol Robinson’s research in native MS to develop its proprietary drug discovery platform, OdyssION™. The platform integrates novel biochemistry techniques, next-generation native mass spectrometry, and custom chemistry, to allow for the interrogation of protein interactions in its native ecosystem while avoiding the confounding complexity of the cell.
This latest publication demonstrates some of the key benefits that OMass’ OdyssION™ platform offers in the search for new drugs against inadequately drugged or previously intractable targets. OMass is advancing a pipeline of small molecule therapeutics in rare diseases and immunological conditions that target GPCRs, solute carriers and complex-bound proteins.
Co-author Ali Jazayeri PhD, Chief Scientific Officer of OMass added: “GPCRs represent one of the most important target class of proteins for drug discovery, with more than 30% of clinically marketed drugs active against this receptor family. But, despite their prevalence as therapeutic targets, challenges remain in drugging these receptors.
We believe our native MS platform, as demonstrated with this latest publication, opens new possibilities to develop drugs against new GPCR targets, as well as other target classes such as solute carriers and complex-bound proteins.”
1. https://www.nature.com/articles/s41557-022-01041-9
-ENDS-
For further information, please contact:
|
OMass Therapeutics
|
Consilium Strategic Communications Sue Charles / Stella Lempidaki / Kumail Waljee |
About OMass Therapeutics
OMass Therapeutics is a biotechnology company discovering medicines against highly-validated target ecosystems, such as membrane proteins or intracellular complexes. The company’s unique OdyssION™ technology platform comprises novel biochemistry techniques, next-generation native mass spectrometry, and custom chemistry. This allows OMass to interrogate not just the target, but also the interaction of the target with its native ecosystem, separate from the confounding complexity of the cell. The result is cell-system fidelity with cell-free precision. OMass is advancing a pipeline of small molecule therapeutics in rare diseases and immunological conditions, that target solute carriers, complex-bound proteins, and GPCRs.
Headquartered in Oxford, UK, OMass has raised over $150M (£119M) from a top-tier international investor syndicate, including Syncona, Oxford Science Enterprises, GV, Northpond Ventures, and Sanofi Ventures.
To learn more, please visit www.omass.com. Follow us on LinkedIn and Twitter.
Company’s medicines target unprecedented mechanisms that free the body’s normal immunity against cancer
Immuno-oncology discovery engine based on ongoing academic-biotech research from the Lieping Chen lab at Yale School of Medicine
First pipeline programs focused on novel mechanisms of tumor T-cell exclusion
Boston, Mass., and West Haven, Conn. – October 25, 2022 – Normunity, Inc., a biotechnology company creating novel precision anti-cancer immunotherapies, today announced its launch and Series A financing of $65 million. The Series A was led by Canaan Ventures and included participation by Sanofi Ventures, Taiho Ventures and Osage University Partners. Normunity’s new class of agents, called immune normalizers, target previously undiscovered mechanisms of immune disruption in cancer. The company’s pipeline is drawn from the ongoing and interactive academic-biotech research taking place at Yale School of Medicine, leveraging proprietary discovery platforms from the lab of Dr. Lieping Chen, an immuno-oncology luminary who identified PD-L1 (B7-H1) among other seminal contributions to the field.
“The normal immune system is a powerful anti-cancer force, and effective immune-stimulating strategies can result in long-lasting cancer remissions, even cures. Yet, today’s immunotherapies don’t capitalize on the full anti-cancer potential of the normal immune system, and most cancer patients do not respond to available treatment options. This leaves an untapped immune biology that we have begun to uncover,” said Lieping Chen, MD, PhD, scientific founder of Normunity and UTC Professor in Cancer Research and Professor of Immunobiology, of Dermatology and of Medicine (Medical Oncology) at Yale School of Medicine. “Working together with Normunity, we are finding previously hidden mechanisms of tumor-dependent immune disruption and we aim to usher in a new era of drug discovery for precision immuno-oncology with medicines that normalize immune function.”
The Series A financing will enable Normunity to advance its emerging pipeline of immune normalizers into the clinic. The company’s initial pipeline programs target mechanisms that drive the exclusion of T cells into immune-sensitive tumors and aim to deliver an active and effective immune system into ‘cold’ tumors. Proceeds from the financing will also be used to build on multiple discovery platforms in the Lieping Chen lab to pursue additional mechanisms that block normal anti-cancer immunity.
“It is so clear to everyone involved in launching Normunity that there is remarkable potential to translate the Lieping Chen lab’s leading-edge discoveries into breakthrough cancer immunotherapies. We have built the right team and the right seamless collaboration between Normunity and the Lieping Chen lab to rapidly cultivate scientific discovery, jointly share expertise, and advance a novel class of drugs that can potentially set a new standard in the immuno-oncology landscape,” said Tim Shannon, MD, General Partner at Canaan Ventures and Chair of Normunity’s Board of Directors.
The company’s unique model is a collaborative and iterative workflow between the Lieping Chen lab and Normunity to identify novel immuno-oncology mechanisms through proprietary platforms, interrogate and validate the emerging targets together pre-clinically and clinically. This academic-biotech alliance leverages world-class scientists with deeply experienced drug designers and developers in a new way.
“Normunity is leading the way in establishing a new roadmap for I-O drug discovery with a simple and powerful premise: to free the immune system to work with maximal potential against cancer. With our scientific approach, we are pioneering novel mechanisms where there is unexplored potential for new medicines. With our R&D model, we have established a first-of-a-kind collaboration with the Lieping Chen lab that has already been prolific in identifying and validating novel targets,” said Rachel Humphrey, MD, founding Chief Executive Officer. “We have strong momentum advancing our pipeline of immune normalizers as we translate groundbreaking biology into life-changing medicines for cancer patients.”
Leadership in immuno-oncology
Leading the academic-biotech teams that are harnessing the potential of the Lieping Chen lab’s proprietary platforms and Normunity’s drug development are Rachel Humphrey, MD, Normunity’s Chief Executive Officer, and Lieping Chen, MD, PhD, whose lab is responsible for many important discoveries in immuno-oncology pathways.
Among his seminal contributions to the field of immuno-oncology, Dr. Chen played an integral role in the discovery of the PD‑1/PD-L1 pathway and its immune-suppressive functions. His lab performed foundational work that led to the invention of anti‑PD‑1/PD‑L1 antibody therapy. In addition, he was deeply involved in the first-in-human trial for Opdivo® (nivolumab), and invented PD-L1 clinical measurement tools. Dr. Humphrey has 25 years of experience with a focus in oncology, including leading the development of the cancer immunotherapies Yervoy® (ipilimumab) at BMS and Imfinzi® (durvalumab) at AstraZeneca, as well as the tyrosine kinase inhibitor Nexavar® (sorafenib) at Bayer. Her biotech leadership roles include CMO at Black Diamond Therapeutics, CytomX and Mirati, and independent Board of Director positions at CytomX, Xilio, Pyxis and Sporos.
The Normunity team brings together experienced drug development leaders with decades of experience in novel drug discovery and clinical advancement of novel cancer and immuno-oncology drugs who have played key roles in the development of more than 30 distinct approved drugs.
About Normunity
Normunity, Inc., is a biotechnology company creating a new class of precision immuno-oncology medicines, called immune normalizers, that target novel mechanisms that free the body’s normal immunity against cancer. Based on an ongoing, interactive academic-biotech partnership with the lab of Dr. Lieping Chen at Yale School of Medicine, Normunity is targeting newly-discovered mechanisms of immune disruption in cancer based on proprietary discovery platforms that elucidate the complex interactions between human cancer and the immune system. The company is advancing an emerging pipeline of immune normalizers, including initial drug programs that target mechanisms that drive the exclusion of T-cells from immune-sensitive tumors and other mechanisms that are barriers to normal immune function in cancer. Normunity is located in Boston, Mass. and West Haven, Conn. For more information, please visit www.normunity.com.
Media Contact
Kathryn Morris, The Yates Network LLC
914-204-6412
kathryn@theyatesnetwork.com
- Founded by leading experts in covalent chemistry, proteomics, and computational sciences from Stanford University and Harvard Medical School
- Series A financing led by Sanofi Ventures with participation from Atlas Venture, Access Biotechnology, Vertex Ventures HC, Digitalis Ventures, Alexandria Venture Investments, and others
Cambridge, Mass., October 14, 2022 – Matchpoint Therapeutics, a biotechnology company founded to leverage the power of covalency for the discovery of precision small molecule medicines, launched today with $100 million in financing. Matchpoint’s funding includes a recently closed $70 million Series A financing led by Sanofi Ventures and joined by Vertex Ventures HC, Digitalis Ventures, Alexandria Venture Investments, other new investors, and all existing investors. The Series A follows a $30 million seed financing round co-led by Atlas Venture and Access Biotechnology in November 2021. The financing will be used to support further advancement of the company’s proprietary platform and its discovery pipeline of novel covalent molecules, initially focused on immunology.
“We created Matchpoint to harness the powerful properties of covalency and enable the targeting of disease-causing proteins otherwise unamenable to small molecule intervention,” said Andre Turenne, President and Chief Executive Officer of Matchpoint. “We believe the immunology space is greatly underserved by current small molecule therapies and are excited by the potential of our proprietary platform to deliver highly specific covalent medicines for several of its most important and difficult targets.”
The durable target engagement achieved with covalent chemistry imparts improved potency, greater selectivity, and lower systemic exposure than otherwise possible. Matchpoint is pursuing genetically or clinically validated targets for which covalent chemistry is the only or best modality for therapeutic intervention.
Matchpoint's Advanced Covalent Exploration (ACE) platform brings together a suite of proprietary tools to revolutionize the discovery and development of covalent medicines:
- Industry-leading chemoproteomics that identifies novel covalent binders to disease- causing proteins, including those with low abundance, in their native cellular environment and provides proteome-wide assessment of selectivity
- Machine learning algorithms that reveal the rules of covalency to guide target prioritization and predict privileged scaffolds to support medicinal chemistry and library design
- Proprietary libraries of covalent compounds that are continuously evolving and informed by predictive algorithms
###
“Our platform maximizes the potential for detecting novel covalent labelers of active sites, allosteric sites, and cryptic sites on targets that have historically proved elusive to drug. It also enables a thorough assessment of a molecule’s selectivity profile early in the discovery process,” said Nathanael Gray, PhD, Matchpoint co-founder and Professor of Chemical and Systems Biology at Stanford University. “The range of applications of this platform is very exciting given that it is as well suited for the discovery of covalent inhibitors as it is for covalent degraders and covalent molecular glues.”
“Much of the proteome that could be targeted by covalent drugs has to date remained invisible to standard chemoproteomics methods. Our platform, combining proprietary chemoproteomics approaches and artificial intelligence, is overcoming this limitation and the word-class team assembled at Matchpoint is uniquely positioned to translate these advances into the next generation of precision covalent medicines,” said Edward Chouchani, PhD, Matchpoint co-founder and Associate Professor of Cell Biology at Harvard Medical School and Dana-Farber Cancer Institute.
In addition to Drs. Gray and Chouchani, Matchpoint’s founders include Dr. Jianwei Che from Dana-Farber Cancer Institute and Dr. Tinghu Zhang from Stanford University. In connection with the financing, Jason Hafler, PhD, Managing Director of Sanofi Ventures, joins Mr. Turenne, Daniel Becker, MD, PhD, Managing Director at Access Biotechnology, and Bruce Booth, DPhil, Partner at Atlas Venture, on the company’s Board of Directors.
"The field of covalent medicine saw a resurgence some 15 years ago that led to several important new medicines, most of which were for oncology indications," said Bruce Booth, DPhil, Chair of Matchpoint’s Board of Directors. “With the latest advances in chemoproteomics, computational sciences, and synthetic chemistry, we now have the opportunity to transform the treatment of autoimmune diseases and other chronic diseases with precision covalent small molecules. We are delighted to welcome Sanofi Ventures and our additional new investors to this pursuit as we continue to build Matchpoint into the leader in covalent medicines.”
About Matchpoint Therapeutics
Matchpoint is a biotechnology company harnessing the power of covalency to discover precision covalent medicines to transform the treatment of immune diseases and other serious illnesses. The company's proprietary Advanced Covalent Exploration (ACE) platform integrates advanced chemoproteomics, machine learning and covalent chemistry library evolution.
Matchpoint has an emerging pipeline of novel covalent medicines initially focused on immunology. For more information, please visit www.matchpointtx.com.
Matchpoint Media Contact: Cory Tromblee Media@MatchpointTx.com
Copenhagen, Denmark and Boston, US, 11th October 2022 – Muna Therapeutics (Muna), a biopharmaceutical company discovering and developing novel therapeutics for the treatment of Parkinson’s disease and other neurodegenerative disorders, announced today that it has been awarded a $4.9 million grant from The Michael J. Fox Foundation for Parkinson’s Research (MJFF).
The grant will be used to support ongoing preclinical research and development of novel, brain- exposed, small molecule potassium channel type 1.3 (Kv1.3) blockers to abrogate neuroinflammation driven by disease-associated microglia and enhance neuroprotection as a disease-modifying therapy for patients with Parkinson’s disease.
Parkinson’s disease is a neurodegenerative disorder associated with motor symptoms (slow movement, tremor, rigidity, walking and imbalance) and a wide variety of non-motor complications (cognitive impairment, mental health disorders, sleep disorders, pain and other sensory disturbances). Motor impairments, such as dyskinesias (involuntary movements) and dystonias (painful involuntary muscle contractions) contribute to limitations in speech, mobility and restrictions in many life areas. More than 10 million people worldwide are estimated to be living with Parkinson’s with no current cure.
Muna is focused on addressing the staggering unmet medical need experienced by Parkinson’s patients around the world with its transformative, therapeutic all-in-human target discovery and validation platform which aims to preserve brain function and enhance resilience to neurodegenerative diseases.
“We are very pleased to receive this significant funding from The Michael J. Fox Foundation to fund this promising research which has the potential to significantly improve the lives of people with Parkinson’s. Kv1.3 plays an important role in creating and maintaining neuroinflammation in Parkinson’s and other neurodegenerative diseases. Reducing neuroinflammation by blocking Kv1.3 has tremendous potential to slow or prevent neurodegeneration and disease progression,” said Rita Balice-Gordon, Chief Executive Officer of Muna Therapeutics.
“The studies will support ongoing medicinal chemistry and structural biology efforts as well as extend understanding of the mechanism of Kv1.3 in microglial activation and the role of Kv1.3 blockade, in vitro in human cells and in vivo in humanized mouse models, to achieve the normalization of disease- associated microglial phenotypes, which will enhance neuroprotection,” said Niels Plath, Chief Scientific Officer of Muna.
"Fostering a robust and healthy pipeline of therapies to improve the lives of people with Parkinson’s is core to MJFF’s mission. We are proud to support the work of researchers at Muna Therapeutics investigating a potential disease modifying therapy for people with Parkinson’s,” said Brian Fiske, PhD, Co-Chief Scientific Officer, MJFF.
The grant from MJFF will support work at Muna Therapeutics and in collaboration with Professor Bart De Strooper at VIB/KU Leuven, a scientific co-founder of Muna, for two years.
For more information please contact:
Optimum Strategic Communications
Mary Clark, Manel Mateus, Zoe Bolt Tel: +44 (0) 20 3882 9621
Email: muna@optimumcomms.com
Muna Therapeutics
Rita Balice-Gordon, CEO
Email: balicegordon@munatherapeutics.com
About Muna Therapeutics
Muna Therapeutics is a private biopharmaceutical company founded in 2020 and based in Copenhagen, Denmark and Leuven, Belgium and the United States. Muna discovers and develops therapies that slow or stop devastating neurodegenerative diseases including Parkinson’s, Alzheimer’s, Frontotemporal Dementia and Multiple Sclerosis. These disorders impact memory, movement, language, behavior and personality resulting in disability and death of millions of patients around the globe. Muna’s groundbreaking science is focused on identifying new medicines to preserve cognition and other brain functions and enhance resilience to neurodegenerative diseases. Our name reflects this focus: Muna means ‘to remember’ in Old Norse. For more information, visit www.munatherapeutics.com
- T-LGLL is an autoimmune disorder characterized by cytotoxic T lymphocytes which attack neutrophils and red blood cell precursors
Newton, Massachusetts, October 4, 2022, Abcuro, Inc., a clinical-stage biotechnology company developing therapies for the treatment of autoimmune diseases and cancer through precise modulation of cytotoxic T and NK cells, today announced initiation of a Phase 1/2 dose escalation trial to evaluate the safety, tolerability, and proof-of-concept of ABC008 in patients with T cell large granular lymphocytic leukemia (T-LGLL) who suffer from anemia and/or neutropenia. ABC008 is a first-in-class anti-KLRG1 antibody capable of selectively depleting highly cytotoxic T cells, while sparing regulatory T cells, central memory T cells, and other immune cells.
T-LGLL is an autoimmune disorder characterized by clonally expanded CD3+ CD8+ cytotoxic T lymphocytes which attack neutrophils and red blood cell precursors, leading to neutropenia and anemia. Neutropenia can lead to frequent infections, a major cause of premature death in patients with T-LGLL, while anemia results in transfusion dependence in approximately one third of patients.
"Initiating the Phase 1/2 clinical trial of ABC008 represents an important step toward a potential breakthrough in the treatment of T-LGLL,” said Jeffrey Wilkins, MD, Chief Medical Officer of Abcuro. “This trial builds upon prior published data demonstrating the ability of ABC008 to deplete highly cytotoxic T cells, which attack and destroy muscle tissue in inclusion body myositis, an autoimmune disease.”
"The limited efficacy of current standard of care is reflected in an overall reduced life expectancy for T- LGLL patients,” said Paul Shami, MD, Professor of Medicine in the Division of Hematology and Hematologic Malignancies at the University of Utah and Principal Investigator. “Without any approved treatment options for T-LGLL, many patients turn to off-label therapies, such as methotrexate, a non- specific immunosuppressant, which often have limited effectiveness.”
About ABC008
ABC008 is a first-in-class anti-KLRG1 antibody capable of selectively depleting highly cytotoxic T cells, while sparing regulatory and central memory T cells. ABC008 has been designed to treat diseases mediated by highly cytotoxic T cells, including the autoimmune muscle disease inclusion body myositis (IBM), T cell large granular lymphocytic leukemia (T-LGLL), and mature T cell malignancies. The US Food and Drug Administration (FDA) has granted Orphan Drug Designation to ABC008 for the treatment of IBM.
About Abcuro
Abcuro is a clinical stage biotechnology company developing first-in-class immunotherapies for the treatment of autoimmune diseases and cancer through precise modulation of highly cytotoxic T and NK cells. The company’s lead program is ABC008, which is currently in clinical trials for inclusion body myositis (IBM). ABC008 is also advancing into clinical trials for T cell large granular lymphocytic leukemia. The company is also developing ABC015 to selectively activate highly cytotoxic T and NK cells for treating cancer. For more information, visit us on LinkedIn and at abcuro.com.
Media Contact:
Christine Quern
CBQ Communications cq@christinequern.com 617.650.8497
- Industry veteran brings extensive experience in building and leading biopharmaceutical companies
Newton, Massachusetts, October 3, 2022, Abcuro, Inc., a clinical-stage biotechnology company developing therapies for the treatment of autoimmune diseases and cancer through precise modulation of cytotoxic T and NK cells, today announced the appointment of Alex Martin as Chief Executive Officer. He brings to Abcuro more than 30 years of leadership experience in the biopharmaceutical industry.
"We are excited to welcome Alex Martin as CEO of Abcuro, as he brings extensive experience in building and leading biopharmaceutical companies as well as a track record of delivering value for patients, the clinical community, and investors,” said John Edwards, Executive Chair of Abcuro. “I look forward to working with Alex as we accelerate Abcuro’s growth and advance our clinical programs in the areas of autoimmune disease and cancer.”
Mr. Martin brings over 30 years of experience building companies and closing transactions in the biopharmaceutical industry. He was most recently Chief Executive Officer of Palladio Biosciences, a clinical stage biopharmaceutical company which was acquired by Centessa Pharmaceuticals (CNTA), and previously served as Chief Executive Officer of Realm Therapeutics (RLM) which was acquired by ESSA Pharma (EPIX). Prior to this, he was the Chief Operating Officer of Intercept Pharmaceuticals (ICPT), and Chief Finance Officer of BioXell (BXLN) which was acquired by Cosmo Pharmaceuticals (COPN). Mr. Martin began his career at SmithKline Beecham Pharmaceuticals where he held roles of increasing responsibility in marketing and business development and later joined Novartis as Vice President, Global Business Development & Licensing. He holds a BA from Cornell University and an MBA from Harvard.
“It’s an honor to join the team at Abcuro, a company built on an unmatched ability to selectively target highly cytotoxic T cells in diseases and conditions with high unmet need, and a strong commitment to delivering novel, life-changing therapies for patients,” said Mr. Martin. “I look forward to working with the highly talented team at Abcuro and building on company’s momentum and promising clinical data from the lead program ABC008, to make positive impact on the lives of patients.”
About ABC008
ABC008 is a first-in-class anti-KLRG1 antibody capable of selectively depleting highly cytotoxic T cells, while sparing regulatory and central memory T cells. ABC008 has been designed to treat diseases mediated by highly cytotoxic T cells, including the autoimmune muscle disease inclusion body myositis (IBM), T cell large granular lymphocytic leukemia (T-LGLL), and other mature T cell malignancies. The US Food and Drug Administration (FDA) has granted Orphan Drug Designation to ABC008 for the treatment of IBM.
About Abcuro
Abcuro is a clinical stage biotechnology company developing first-in-class immunotherapies for the treatment of autoimmune diseases and cancer through precise modulation of highly cytotoxic T and NK cells. The company’s lead program is ABC008, which is currently in clinical trials for inclusion body myositis (IBM). ABC008 is also advancing into clinical trials for additional autoimmune disease indications. The company is also developing ABC015 to selectively activate highly cytotoxic T and NK cells for treating cancer. For more information, visit us on LinkedIn and at abcuro.com.
Media Contact:
Christine Quern
CBQ Communications cq@christinequern.com 617.650.8497
- NT-0796 successfully completes Phase 1 study, demonstrating blood-brain barrier penetration and reduced inflammatory biomarkers supporting advancement in neuroinflammatory and peripheral inflammatory diseases
- Second clinical candidate NT-0249 achieves positive interim results from its Phase 1 study supporting once daily dosing for peripheral inflammatory disease
LEXINGTON, MA – September 21, 2022 – NodThera, a clinical-stage biotechnology company developing a new class of potent and selective oral, small molecule NLRP3 inflammasome inhibitors to treat diseases driven by chronic inflammation, today announced positive Phase 1 clinical readouts for its first and second clinical candidates, NT-0796 and NT-0249. NT-0796 has completed its Phase 1 study confirming brain penetration with excellent pharmacokinetic (PK) & pharmacodynamic (PD) profiles and NT-0249 has completed dosing of the Phase 1 single ascending dose cohorts confirming a potentially best-in-class PK/PD profile and the potential for once-a-day dosing. The results collectively support further development and clinical evaluation in a range of CNS and peripheral inflammatory diseases.
“We are delighted that NT-0796 continues to demonstrate an exceptional and differentiated clinical profile,” said Adam Keeney, Chief Executive Officer at NodThera. “Neurological and neurodegenerative diseases are a significant and growing burden to patients and society. This is the first clinical demonstration of a brain penetrant inhibitor of the NLRP3 inflammasome, which represents a major milestone for addressing this urgent medical challenge.”
NT-0796 is a novel chemotype, designed as an orally bioavailable, brain penetrant NLRP3 inhibitor. The Phase 1 study showed exposures of NT-0796 and its bioactive metabolite NDT-19795 increased linearly with dose. A dose-dependent pharmacodynamic effect was also observed through inhibition of stimulated IL-1β and IL-18 in ex vivo blood samples, which translated into an anti-inflammatory effect via reduction of key inflammatory biomarkers, including C-reactive protein (CRP). Blood-brain barrier penetration of NT-0796 was verified with cerebrospinal fluid (CSF) drug concentrations in excess of anti-inflammatory free blood concentrations. Overall, NT-0796 was safe and well tolerated and no drug-related liver function test (LFT) abnormalities were observed.
NodThera’s second clinical compound, NT-0249, is a peripherally restricted NLRP3 inflammasome inhibitor that has successfully completed its Phase 1 single ascending dose study. NT-0249 was safe and well tolerated with proportional increases in drug exposure with increasing dose. This profile has the potential to be best-in-class as a peripheral NLRP3 inflammasome inhibitor, demonstrating pharmacokinetics consistent with a once-a-day therapy and pharmacodynamics confirming a low clinical dose for efficacy.
“From design to development, the success of our Phase 1 data represents a breakthrough in targeting the NLRP3 inflammasome both peripherally and in the CNS” said Alan Watt, NodThera’s CSO and President of R&D. “Delivery of NT-0796 into the CNS and the positive interim data for NT-0249 continues to reinforce NodThera’s leadership in the NLRP3 inflammasome field.”
About NodThera
NodThera is a clinical-stage biotechnology company developing a new class of potent and selective NLRP3 inflammasome inhibitors for the treatment of diseases driven by chronic inflammation. Led by an experienced management team, NodThera is leveraging new insights into inflammasome biology and chemistry to build a clinically advanced portfolio of highly differentiated small molecule NLRP3 inflammasome inhibitors. The company was founded in 2016 by Epidarex Capital and financed by 5AM Ventures, Cowen Healthcare Investments, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Lexington, MA, with additional locations in Cambridge, UK and Seattle, WA. Learn more at www.nodthera.com or follow us on LinkedIn.
Media Contact
Darby Pearson
Verge Scientific Communications
- Lead candidate NT-0796 achieves positive interim results from Phase 1 study, supporting further progression for the treatment of a range of inflammatory diseases
- Second lead candidate NT-0249 advances into first-in-human Phase 1 study to enable clinical development in peripheral chronic inflammatory disease
- Third candidate NT-0527 is announced as a novel brain-penetrant NLRP3 inflammasome inhibitor advancing through IND-enabling studies-
LEXINGTON, MA – May 10, 2022 – NodThera, a clinical-stage biotechnology company developing a new class of potent and selective oral, small molecule NLRP3 inflammasome inhibitors to treat diseases driven by chronic inflammation, today announced several key advancements across the portfolio. NodThera’s lead candidate, NT-0796, demonstrated positive interim results from its Phase 1 single-ascending dose (SAD) study. Additionally, the company has commenced first-in-human dosing in the Phase 1 study of its second lead candidate, NT-0249, and announced the selection of its third pipeline candidate, NT-0527 – a brain- penetrant NLRP3 inflammasome inhibitor from a novel chemotype.
The positive interim results from the SAD portion of the Phase 1 trial with NT-0796 represent early clinical proof-of-mechanism for NT-0796 as a potent NLRP3 inflammasome inhibitor. Across all dosing cohorts, NT-0796 was safe and well tolerated and shown to be orally bioavailable with a dose-proportional pharmacokinetic (PK) profile. This portion of the study also showed a dose-dependent pharmacodynamic (PD) effect through the ability to lower IL-1β and IL-18 levels in an ex vivo NLRP3-stimulation assay. These results confirm the criteria to advance NT-0796 further in development and continue the ongoing multiple- ascending dose (MAD) portion of the Phase 1 study to assess brain exposure through cerebrospinal fluid (CSF) sampling.
“NT-0796 has demonstrated robust proof of mechanism and translation from preclinical studies to humans, both validating and further de-risking the development of NT-0796 as a potentially best-in-class, oral, small molecule NLRP3 inflammasome inhibitor,” said NodThera’s Chief Executive Officer, Adam Keeney. “We are encouraged by these first-in-human results as we work to progress NT-0796 in inflammatory diseases impacting millions of patients, many with limited to no treatment options.”
Building on successful clinical progress with NT-0796, NodThera has also initiated dosing of the first healthy volunteers in the Phase 1 trial of the company’s second lead candidate, NT-0249. NT-0249 is a potent inhibitor of the NLRP3 inflammasome with favorable development characteristics supporting advancement to treat chronic inflammatory diseases of the body. The primary objective of this study will be to assess the safety and tolerability of NT-0249, with secondary objectives to assess the PK and PD (ability to lower IL- 1β and IL-18 levels) after single and multiple ascending doses.
In addition to advancing two novel candidates into clinical trials, NodThera is further expanding its diverse portfolio with the announcement of NT-0527 as the third oral small molecule NLRP3 inflammasome inhibitor from a novel chemotype to be added to its pipeline. NT-0527 is uniquely designed to inhibit the NLRP3 inflammasome in the brain, with potential to treat a broad range of neuroinflammatory diseases.
“Each of NodThera’s three portfolio candidates feature unique chemotypes that are distinct from one another, offering specific benefits that can be collectively used to cover a range of inflammatory diseases of the brain and body” shares NodThera’s Chief Scientific Officer, Alan Watt. “With the recent addition of NT-0527 as the third candidate in our growing pipeline, NodThera is now advancing novel CNS-penetrant and peripherally-restricted NLRP3 inflammasome inhibitors with differentiated chemistry unlike any other portfolio in the field.”
About NodThera
NodThera is a clinical-stage biotechnology company developing a new class of potent and selective NLRP3 inflammasome inhibitors for the treatment of diseases driven by chronic inflammation. Led by an experienced management team, NodThera is leveraging new insights into inflammasome biology and chemistry to build a portfolio of highly differentiated small molecule NLRP3 inflammasome inhibitors. The company was founded in 2016 by Epidarex Capital and financed by 5AM Ventures, Cowen Healthcare Investments, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Lexington, MA, with additional locations in Cambridge, UK and Seattle, WA. Learn more at www.nodthera.com or follow us on LinkedIn.
Media Contact
Darby Pearson
Verge Scientific Communications
dpearson@vergescientific.com
- Financing led by new investors, GV, Northpond Ventures and Sanofi Ventures, with existing investors, Syncona, Oxford Science Enterprises, and Oxford University also participating
- Investment will advance OMass’s portfolio of novel drugs against highly-validated, yet intractable or inadequately drugged, membrane- and complex-bound proteins towards the clinic
- OMass’s pipeline of small molecule therapeutics includes five programs in rare diseases and immunological conditions that target solute carriers, complex-bound proteins and GPCRs, all discovered using OMass’s proprietary OdyssION™ platform
Oxford, United Kingdom – 28 April 2022 – OMass Therapeutics (‘OMass’, or ‘the Company’), a biotechnology company that identifies medicines against highly validated target ecosystems, today announces its $100 million (£75.5 million) Series B financing round. The international syndicate of top-tier life science specialists was led by new investors, GV, Northpond Ventures and Sanofi Ventures, with existing investors, Syncona, Oxford Science Enterprises and Oxford University also participating.
Proceeds from the financing will be used to advance OMass’s portfolio towards clinical trials. This includes the development of an insurmountable antagonist of the MC2 receptor for congenital adrenal hyperplasia, a gasdermin D inhibitor for the treatment of inflammatory diseases, a GPR65 agonist for the treatment of inflammatory bowel disease and two earlier-stage programs targeting solute carriers.
Ros Deegan, CEO of OMass, said: “The completion of this oversubscribed round with such high-calibre investors is recognition of the significance of our OdyssION™ platform and its potential to support the development of an exciting portfolio of novel drug candidates. We have already made significant progress against highly validated but previously ‘undruggable’ targets and can now accelerate them towards clinical development while continuing to expand our pipeline.”
Originally spun out of Oxford University, OMass has commercialised Professor Dame Carol Robinson’s breakthrough research in native mass spectrometry to develop its proprietary drug discovery platform, OdyssION™. The platform integrates novel biochemistry techniques, next-generation native mass spectrometry, and custom chemistry, to allow for the interrogation of protein interactions within its native ecosystem while avoiding the confounding complexity of the cell.
OMass’s OdyssION™ platform delivers several key benefits in the search for new drugs against previously intractable targets, including discovering drug binders with high sensitivity without filtering on activity, establishing an unambiguous link between binding and function to drive smart lead optimization, and identifying natural allosteric sites that can be targeted for drug discovery.
Following the financing, Scott Biller, Ph.D., Executive Venture Partner at GV, Diana Bernstein, Ph.D., Vice President at Northpond Ventures, and Laia Crespo, Ph.D., Partner at Sanofi Ventures, will join the OMass Board of Directors.
Scott Biller, Ph.D., Executive Venture Partner at GV, said: “By studying proteins in their native state as found within the cell, the OMass platform can address many important targets that have been elusive until now. Through this platform, OMass has created an impressive portfolio of therapeutic programs with the potential to dramatically improve the lives of patients.”
Diana Bernstein, Ph.D., Vice President at Northpond Ventures, said: “OMass is fundamentally shifting the process by which we identify and evaluate chemistry for the most challenging drug targets. Native mass spectrometry uniquely permits simultaneous evaluation of drug binding and functional effect, and OMass is a leader in this pursuit.”
Laia Crespo, Ph.D., Partner at Sanofi Ventures, said: “OMass’s vision and the potential of its platform aligns with the purpose of our fund. As a strategic venture investor, we support top tier life science entrepreneurs with innovative ideas and transformative new products and technologies of strategic interest to Sanofi. We partner with management to advance innovation that has the potential to deliver new approaches that can transform patients’ lives and we look forward to supporting OMass as it develops new drugs for the treatment of rare diseases and immunological conditions.”
Edward Hodgkin, Ph.D., Chair of OMass’s Board of Directors and Partner at Syncona added: “We are pleased with this financing round which will support OMass as it looks to progress its pipeline of small molecule drugs. The strength of this global group of top tier life science investors reflects confidence in the company’s technology and supports our ambition to build a sustainable therapeutics business that has the potential to develop novel drugs in areas of high unmet medical need. We are also delighted to welcome Scott Biller, Diana Bernstein and Laia Crespo to the OMass Board, all who have significant experience in transforming technology platforms into sustainable drug discovery and development businesses.”
For further information, please contact:
OMass Therapeutics
Rosamond Deegan, Chief Executive Officer
Phone: +44 (0) 1235 527589
Email: ros.deegan@omass.com
Consilium Strategic Communications
Sue Charles / Chris Gardner / Kumail Waljee
Phone: +44 (0)20 3709 5700
Email: omass@consilium-comms.com
About OMass Therapeutics
OMass Therapeutics is a biotechnology company discovering medicines against highly-validated target ecosystems, such as membrane proteins or intracellular complexes. The company’s unique OdyssION™ technology platform comprises novel biochemistry techniques, next-generation native mass spectrometry, and custom chemistry. This allows OMass to interrogate not just the target, but also the interaction of the target with its native ecosystem, separate from the confounding complexity of the cell. The result is cell-system fidelity with cell-free precision. OMass is advancing a pipeline of small molecule therapeutics in rare diseases and immunological conditions, therapeutics that target solute carriers, complex-bound proteins, and GPCRs.
Headquartered in Oxford, UK, OMass has raised over $150M (£119M) from a top-tier international investor syndicate, including Syncona, Oxford Science Enterprises, GV, Northpond Ventures, and Sanofi Ventures.
To learn more, please visit www.omass.com. Follow us on LinkedIn and Twitter.
About GV
GV provides venture capital funding to bold new companies. Across the fields of life science, enterprise technology, consumer products and services, and frontier technology, GV's portfolio companies aim to improve lives and transform industries. GV's team of world-class engineers, designers, physicians, scientists, marketers, and investors work together to provide startups exceptional support.
Launched as Google Ventures in 2009, GV is the venture capital arm of Alphabet, Inc. GV helps startups interface with Google, providing unique access to the world’s best technology and talent.
GV has over $8 billion under management. Notable investment outcomes include Uber, Slack, One Medical Group, Nest, Flatiron Health, and Duo Security. GV is headquartered in Mountain View, California, with offices in San Francisco, Boston, New York, and London.
Find out more: www.gv.com
About Northpond Ventures
Northpond Ventures is a multi-billion dollar science-driven venture capital firm based in Cambridge, MA; San Francisco, CA; and Bethesda, MD. Northpond has been named one of the three most active lead biotech investors in 2021 by Crunchbase, and the most active lead investor in life science solutions and molecular diagnostics by Silicon Valley Bank. It is particularly engaged in the research ecosystem, having founded a bioengineering laboratory at Harvard, and sponsored a prize for women entrepreneurs at MIT. It has led over 50 financings over the past several years, with a high percentage having an academic origin.
Learn more at www.npv.vc
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi. Sanofi Ventures invests in early-stage biotech and digital health companies with innovative ideas and transformative new products and technologies of strategic interest to Sanofi. Among these areas are oncology, immunology, rare diseases, vaccines, potential cures in other core areas of Sanofi’s business footprint, and digital health solutions.
Find out more: www.sanofiventures.com
About Syncona
Syncona's purpose is to invest to extend and enhance human life. We do this by founding and building companies to deliver transformational treatments to patients in areas of high unmet need.
Our strategy is to found, build and fund companies around exceptional science to create a diversified portfolio of 15-20 globally leading healthcare businesses for the benefit of all our stakeholders. We focus on developing treatments for patients by working in close partnership with world-class academic founders and management teams. Our balance sheet underpins our strategy enabling us to take a long-term view as we look to improve the lives of patients with no or poor treatment options, build sustainable life science companies and deliver strong risk-adjusted returns to shareholders.
Find out more: www.synconaltd.com
About Oxford Science Enterprises
Oxford Science Enterprises is a Science Business Builder, committed to helping solve the world’s toughest problems for more people, in more places, faster. The company does this by transforming world-leading science into world-changing businesses, partnering the best scientists from the world’s best university with the best business brains. Oxford Science Enterprises grows its companies with care and expertise, investing for real-world impact, not only financial returns, and re-investing proceeds back into the next generation of original research and world-changing businesses.
Since 2015, the company has received an automatic stake in all Oxford University science spinouts and has taken a leading role in creating and building enterprises that address problems that affect people in life-changing ways: their health, the availability of food, the survival of the planet.
Find out more: oxfordscienceenterprises.com | Twitter | LinkedIn
Led by Section 32 and Sanofi Ventures, funding will enable Nucleai to expand its commercial footprint in biopharma and further develop its state-of-the-art spatial biology platform.
TEL AVIV, Israel — March 22, 2022 — Nucleai, an AI-powered spatial biology company with a mission to transform drug development and clinical treatment decisions by unlocking the power of pathology data, today announced that it had closed a $33 million Series B financing round, jointly led by Section 32 and Sanofi Ventures. Andy Harrison, Managing Partner at Section 32, and Cris De Luca, Global Head, Digital Investments at Sanofi Ventures, will join Nucleai’s Board of Directors. Section 32 Managing Partner Michael Pellini, MD, will join Nucleai’s Board as an observer. Nucleai plans to use the new funding to further develop its platform and expand its commercial footprint across biopharmaceutical companies and contract research organizations (CROs), who are applying its technology throughout translational research, clinical trials, and novel applications for drug discovery.
Existing investors, including Debiopharm, Fosun, Vertex, and Grove ventures, participated in this round. This Series B financing brings Nucleai’s overall funding to close to $50 million since the company was founded four years ago.
“Nucleai’s vision is to bring spatial biology to the forefront of precision medicine and to embed the use of our platform in every clinical trial involving tissue over the next few years.” said Avi Veidman, CEO of Nucleai. “We are pleased to bring world-class investors who share our passion and vision to transform drug development and clinical treatment decisions by combining artificial intelligence, big data, spatial biology, and a comprehensive software platform.”
Andy Harrison commented, “Mapping biological microenvironments with spatial mapping technology is an exciting area of discovery that is paving the way for innovative new therapies and diagnostic tools. Nucleai has built a platform that makes spatial analysis scalable and operational, enabling the next generation of actionable insights from massive pathology data sets that have not been analyzed to their fullest potential and could provide significant value to pharmaceutical companies and diagnostic labs.”
“The rapid growth and achievements of the Nucleai team have them well-positioned to be spatial biology platform leaders, transforming current approaches to pathology,” said Cris De Luca. "By harnessing artificial intelligence with spatial data and other data modalities, Nucleai is enabling researchers and clinicians to make better treatment decisions for patients based on a comprehensive, holistic view of cellular locations, interactions, and the tumor microenvironment.”
Nucleai is working with most of the leading pharmaceutical companies to harness spatial biology for new drug development, clinical trials, and clinical treatment decisions. Nucleai’s platform is leveraged for retrospective and prospective patient stratification analysis in clinical trials, driving improvement of the probability of success and improved patient outcomes. Nucleai delivers a comprehensive solution that brings the computational power and scales needed to discover novel biomarkers, predicts patient response with higher-quality predictive biomarkers, identifies new targets, and develops the next generation of pathology-based companion diagnostics.
For more information, go to www.nucleai.ai.
About Section 32
Section 32 is a venture capital fund investing at the frontiers of technology and healthcare. Founded by Bill Maris, the team has vast experience building iconic companies. Our goal is to improve the human condition by accelerating the discovery, development, and distribution of important technologies and life saving medicines. We invest across the entirety of technology and healthcare. To learn more, go to: www.section32.com
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi. Sanofi Ventures invests in early- stage biotech and digital health companies with innovative ideas and transformative new products and technologies of strategic interest to Sanofi. Among these areas are oncology, immunology, rare diseases, vaccines, potential cures in other core areas of Sanofi’s business footprint, and digital health solutions. www.sanofiventures.com
About Nucleai
Nucleai is an AI-powered spatial biology company with a mission to transform drug development and clinical treatment decisions by unlocking the power of pathology data. Nucleai provides pharmaceutical companies, contract research organizations, and diagnostics laboratories with a state-of-the-art AI platform to improve clinical trials and clinical decision-making. For more information, please visit www.nucleai.ai.
# # #
Media Contact
Anthony Petrucci
Bioscribe
anthony@bioscribe.com
512-581-5442
- Fidelity leads round to help Omada deliver on its mission to improve health outcomes for people living with chronic conditions
- Strategic financing will accelerate growth as the company expands its executive team and sees record market adoption
SAN FRANCISCO -- February 23, 2022 -- Omada Health, a chronic care integrated healthcare provider, announced its $192 million Series E funding round led by Fidelity Management & Research Company with participation from aMoon, existing investors Perceptive Advisors, Wellington Management, Civilization Ventures, and others. The company also announced record growth and an expansion of its executive team and Board of Directors with the appointment of Taylor Harris as a new Board member, Nancy Vitale as Chief People Officer and Sunil Kayiti as Chief Technology Officer.
The healthcare industry has reached a tipping point when it comes to chronic disease. There are 96 million U.S. adults diagnosed with pre- diabetes and 37.3 million diagnosed with diabetes, 47% of U.S. adults have hypertension and more than 50% struggle with musculoskeletal issues. The time is now for Omada Health’s multi-product platform to address these conditions through its proven, virtual care approach. Omada has been uniquely successful in helping people navigate the complexity of their chronic conditions by focusing on behavior change and insights-driven coaching, across critical clinical areas for employers and health plans.
“Omada Health’s mission is to solve the problem of chronic care treatment in the U.S. There are so many inefficiencies in the healthcare system that hinder individuals from truly managing their health,” said Sean Duffy, co- founder and CEO of Omada Health. “With a 10-year track record, Omada’s approach is delivering outcomes that outpace the industry. This latest round of funding not only validates, but accelerates our mission in offering virtual chronic care treatment to millions of people across the U.S. With the support of our investors, Fidelity, aMoon, Civilization Ventures and others, we’re able to better support our customer growth and usher in a new model of care.”
This round of funding comes on the heels of record growth for the company. "Omada Health currently serves 1,700+ customers and 550,000 members – up from 1,000 customers in 2019. Omada Health now has access to over 18 million covered lives across our employer and health plan channels, with more than 3.5 million covered lives added through new deals in 2021" reported CFO Steve Cook. Multi-product contracts accounted for 32% of
Omada’s new business deals in 2021. The digital health company also earned preferred status on the Evernorth Digital Health Formulary for its cardiometabolic digital chronic care programs. The preferred status expands Omada Health’s reach to more than 100 million members.
Omada Health will use the $192 million towards the following:
- Accelerate hiring at all levels to meet growing customer demands
- Accelerate technological roadmap for care and coaching personalization to further improve on delivering better health outcomes
- Increased investment in the Omada Insights Lab to unearth the most innovative, cost-effective interventions
“Omada Health’s dedication to health outcomes and mindset-driven care support was a major differentiator that stood out to aMoon in a crowded digital health landscape.” said Tomer Berkovitz, general partner at aMoon Fund. “This latest funding round emphasizes our confidence in the Omada team and our strong belief in the company’s platform as a proven solution across multiple chronic conditions.”
To register for Omada Health’s Mindset 2022 Summit, please click here.
Media Contact
Ali Nix
omadahealth@highwirepr.com
(339) 227-0583
• Completed enrolment of all pregnant women in a Phase 2 study in South Africa and Uganda
• Following clearance from the US FDA, Danish Medicines Agency and UK MRHA, started enrolment of pregnant women in a Phase 2 study in Denmark and the UK
• Completed enrolment of a Phase 1 booster study in the UK
Copenhagen, Denmark, 14 February 2022 – MinervaX, a privately held Danish biotechnology company developing a novel vaccine against Group B Streptococcus (GBS), today announces significant
clinical progress on its maternal GBS vaccine.
MinervaX is developing a maternal vaccine for the prevention of adverse pregnancy outcomes and life- threating infections caused by Group B streptococcus . GBS is responsible for nearly half of all life-threatening infections in newborns during the first 3 months of life as well as a portion of late-term abortions, premature deliveries, or stillbirths during pregnancy. As current preventive strategies are insufficient, there is an urgent need for a maternal vaccine to reduce the burden of GBS globally. MinervaX’s maternal GBS vaccine is based on adjuvanted protein antigens covering close to 100% of clinical GBS isolates.
MinervaX announces today that the enrolment of all 200 pregnant women in a Phase 2 study in South Africa and Uganda has been completed. Furthermore, all babies in the South Africa study population have been delivered and no product related serious adverse events have been reported to date. The study is sponsored by the European Developing Countries Trial Partnership and is listed on clinicaltrials.gov under NCT04596878.
Furthermore, MinervaX announces today that it has started enrolment of 270 pregnant women in a Phase 2 study in Denmark and the United Kingdom. The study is run under an open US FDA IND and is listed on clinicaltrials.gov under NCT05154578.
Finally, MinervaX has also completed dosing healthy adult women previously receiving the company’s GBS vaccine in a Phase 1 booster study in the United Kingdom. The study is listed on clinicaltrials.gov under NCT05005247.
Commenting on the announcement, Per Fischer, Chief Executive Officer of MinervaX, said: “We are very pleased to have completed enrolment of all subjects in our Phase 2 study in Africa, and to have received positive feedback from the US FDA resulting in IND clearance and enrolment for the Phase 2 in Denmark and the UK, and to have completed dosing of the Phase 1 booster study in the UK. The vaccine has, to date, demonstrated high immune responses even in individuals with low levels of preexisting immunity to GBS who are most at risk of invasive disease. The vaccine has also demonstrated a very promising safety profile in both non-pregnant and pregnant women in past and ongoing clinical studies. The progress represents a significant advancement towards initiating Phase 3 clinical studies.”
Gerd Zettlmeissl, Chairman of MinervaX Board of Directors said: “I would also like to congratulate, on behalf of my Board colleagues, the MinervaX team for this outstanding progress towards a potential future GBS vaccine. We are convinced that our vaccine candidate could provide a major global contribution to the health of pregnant women and newborns.”
Enquiries
For more information on MinervaX, please contact:
Per Fischer, CEO: pbf@minervax.com
About MinervaX
MinervaX is a Danish biotechnology company, established in 2010 to develop a prophylactic vaccine against Group B Streptococcus (GBS), based on research from Lund University. MinervaX is developing a GBS vaccine for maternal immunization, likely to have superior characteristics compared with other GBS vaccine candidates in development. The latter are based on traditional capsular polysaccharide (CPS) conjugate technology. By contrast, MinervaX’s vaccine is a protein-only vaccine based on fusions of highly immunogenic and protective protein domains from selected surface proteins of GBS (the Alpha-like protein family). Given the broad distribution of proteins contained in the vaccine on GBS strains globally, it is expected that MinervaX’s vaccine will confer
protection against virtually 100% of all GBS isolates. www.minervax.com
About Group B Streptococcus (GBS)
GBS is responsible for nearly 50% of all life-threatening infections in newborns. At any given time, some 15- 25% of women are spontaneously colonized with GBS, and they run the risk of transmitting the bacteria to their child in the womb, during birth and/or during the first months of life. GBS colonization may lead to late abortions, premature delivery, or stillbirth and, in the newborn child, may result in sepsis, pneumonia or meningitis, all of which carry a significant risk of severe morbidity, long- term disability or death.
Currently, the only preventative strategy available involves the use of intravenously delivered prophylactic antibiotics which does not comprehensively prevent GBS infection in utero or protect against late-onset infection in newborns. Not only is this approach expensive and logistically challenging, it fails to cover all, including the most severe cases in the US and Europe, and is rarely available in resource- limited settings.
The development of a GBS vaccine is also endorsed by Group B Strep Support and Group B Strep International, and GBS has been prioritized by a number of public health organizations. Both increased uptake of immunization among pregnant women and greater awareness of the implications of G S suggest that a safe and effective vaccine targeting GBS would be well suited to address this unmet need.

Phase 1 Ready Program Targeting mTORC1 to be Pursued for Potential Use in Autosomal Dominant Polycystic Kidney Disease and other Indications
CAMBRIDGE, Mass., February 2, 2022 –Today, privately held Navitor Pharmaceuticals, LLC (“Navitor”), announced that Janssen Pharmaceuticals, Inc. (“Janssen”), one of the Janssen Pharmaceutical Companies of Johnson & Johnson has acquired Anakuria Therapeutics, Inc., (“Anakuria”), a company recently formed by Navitor to advance its novel class of selective rapamycin analog mTORC1 inhibitors. Anakuria’s lead Phase 1 ready program, AT-20494 provides Janssen with a first-in-class opportunity in autosomal dominant polycystic kidney disease, or ADPKD. This deal was facilitated by Johnson & Johnson Innovation.
Under the terms of the agreement, Janssen has acquired all outstanding shares of Anakuria, which is now a wholly owned subsidiary of Janssen.
Tom Hughes, president and chief executive officer of Navitor Pharmaceuticals LLC commented, "We are thrilled that the potential value and substantially differentiated profile of Anakuria’s mTORC1 inhibitor program can be explored with Janssen. With decades of experience in developing, manufacturing and commercializing innovative therapies for patients suffering from a broad range of diseases and conditions, Janssen is ideally positioned to rapidly advance our program in the clinic. We also are very pleased that the program has come full circle within the Johnson & Johnson Family of Companies: Johnson & Johnson Innovation – JJDC, Inc., the strategic venture arm of Johnson & Johnson, was one of Navitor’s founding investors and the company also was one of the initial startups incubated in JLABS @ LabCentral, Cambridge, MA.”
Goodwin Proctor LLP acted as legal advisor to Navitor on this transaction.
About AT-20494
AT-20494 is an orally bioavailable small molecule that selectively inhibits activity of mTORC1, the master modulator of cellular metabolism, which is overactive in multiple chronic diseases including autosomal dominant polycystic kidney diseases. AT-20494 is a member of a novel class of rapamycin analogs discovered by Navitor scientists, and will be the first fully selective inhibitor of mTORC1 to be studied in humans. Preclinical studies of AT-20494 have shown that it reduces the burden of cysts and kidney volume in mice carrying mutations in the PKD1 gene, and also reduces signatures of fibrosis and inflammation upon chronic administration.
About mTORC1
Complex 1 of the mechanistic target of rapamycin (mTORC1) activity governs the pace and ability of the cell to synthesize protein and other cellular components. Increased mTORC1 activity contributes to a broad array of diseases of aging by increasing protein misfolding and driving cellular stress, inflammation, and fibrosis.
About Navitor
Navitor Pharmaceuticals, LLC, the parent company for Navitor Pharmaceuticals, Inc., is the leader in the development of mTORC1-targeted therapeutics designed to help patients live longer and healthier lives. The Company’s proprietary platform enables highly specific modulation of mTORC1, the gatekeeper of cellular metabolism and renewal, and has produced two clinical candidates. NV-5138, Navitor’s small molecule that directly activates mTORC1, is in Phase 2 development for treatment-resistant depression, with additional opportunities in cognition and memory, in partnership with Supernus Pharmaceuticals, Inc.
Navitor Media Contact
Jamie Palme (857) 272-0959
jpalme@navitorpharma.com
Collaboration will focus on co-development of unique datasets to understand health in everyday life
January 10, 2022 06:00 AM Eastern Standard Time
SAN MATEO, Calif.--(BUSINESS WIRE)--Evidation, the company creating new ways to measure and improve health in everyday life, is expanding its decade-long collaboration with Sanofi to build upon their joint real-world data initiatives. This new phase will focus on the co-development of unique datasets to develop and validate new measures of health and wellness.
Evidation’s collaboration with Sanofi has delivered groundbreaking results to date, with over 20 studies conducted across 10 therapeutic areas, including diabetes and Type 2 Inflammation, more than 500,000 patients reached, and four studies published. This continued collaboration will further the work Evidation and Sanofi have pioneered to translate person-generated health data into quantified clinical and economic outcomes, a key priority for both companies.
“After nearly a decade of working with Sanofi, we are proud to expand this collaboration agreement to advance the role that real-world data and analysis can play in better understanding health and disease,” said Christine Lemke, Evidation co-founder and co-CEO. “Sanofi has led the way in garnering insights from real-world data in R&D and we’re excited to advance our work together into its next decade."
Sanofi and Evidation announced a prior expansion of their work together in 2017, in addition to Sanofi’s investment in Evidation in the same year.
“Real-world evidence is critical to help us better understand the patient’s health and wellness journey outside of traditional healthcare visits,” said Arnaud Robert, Executive Vice President, Chief Digital Officer, Sanofi. “Through our expanded collaboration with Evidation, we can further our ambition to transform the practice of medicine by connecting more closely with patients and citizens, expanding our geographic capabilities, and increasing diversity to better represent the global population.”
This announcement comes as biopharmaceutical companies, regulators, and payers are working to develop new guidelines on how real-world data should be incorporated into the development and approval of therapeutics. Evidation and Sanofi will continue to contribute to this conversation through similar industry-leading research.
The Evidation network is made up of more than 4.4 million individuals across all 50 states, representing 9 out of every 10 U.S. ZIP codes, allowing organizations like Sanofi access to a highly engaged, diverse population and privacy-conscious research platform.
ABOUT EVIDATION
Evidation measures health in everyday life and enables anyone to participate in ground-breaking research and health programs. Built upon a foundation of user privacy and control over permissioned health data, Evidation’s platform is trusted by millions of individuals—generating data with unprecedented speed, scale, and rigor. We partner with leading healthcare companies to understand health and disease outside the clinic walls. Guided by our mission to enable and empower everyone to participate in better health outcomes, Evidation is working to bring people individualized, proactive, and accessible healthcare—faster. Founded in 2012, Evidation is headquartered in California with additional offices around the globe. To learn more, visit evidation.com, or follow us on Twitter @evidation.
Contacts
MEDIA CONTACT
Matt Miller
press@evidation.com
Dr. Suliman brings over 25 years of drug development, deal-making and company building expertise as ReCode continues advancing its pipeline of novel genetic medicines using its powerful selective organ targeting (SORT) LNP delivery platform
Menlo Park, Calif. And Dallas, Texas – January 10, 2022 – ReCode Therapeutics (the “Company”), a biopharmaceutical company pioneering disease-modifying genetic medicines using its proprietary LNP delivery platform, today announced the appointment of Shehnaaz Suliman, M.D., MBA, M.Phil., as chief executive officer and as a member of the board of directors effective today. Former CEO, David Lockhart, Ph.D., will transition to the role of chief scientific officer and will remain president and a member of the board of directors.
“I am excited about furthering the development of transformative therapeutics for patients with ReCode’s first-in-class LNP platform and promising genetic medicines pipeline,” said Shehnaaz Suliman, M.D., MBA, M.Phil., chief executive officer, ReCode Therapeutics. “There is vast untapped potential in RNA medicines and the delivery of genetic payloads to modify and potentially remedy a wide variety of life-limiting diseases. I am humbled to lead ReCode into its next stage of growth and look forward to partnering with Dr. Lockhart and the team to continue to execute our mission to develop a new generation of first-in-class mRNA-based and gene editing therapies for patients.”
“Dr. Suliman has an exceptional track record of building and transforming small and large life science companies into leading biotechnology companies. Her impressive business acumen, ability to advance assets through the clinic and competency in deal-making make her an outstanding fit for CEO as we enter our next chapter of growth,” said David Lockhart, Ph.D., president and chief scientific officer, ReCode Therapeutics. “I am personally very excited to partner with Dr. Suliman, and we are invigorated by her arrival and look forward to leveraging her expertise as we advance our powerful LNP platform and robust pipeline of treatments for patients with life-limiting genetic diseases.”
Dr. Suliman is a physician, drug developer and dealmaker with over 25 years of experience building and transforming small and large biopharmaceutical companies. She most recently served as president and chief operating officer of Alector, a leading immuno-neurology and immuno-oncology company where she played a critical role growing, scaling, raising capital and advancing a late-stage development pipeline. Prior to Alector, she served as senior vice president, corporate development and strategy at Theravance Biopharma from 2017 to 2019, where she helped deliver a $1B profit sharing partnership with Janssen for the company’s lead JAK inhibitor program. Prior to Theravance, Dr. Suliman was vice president and global head, immunology, infectious diseases and specialty care at Roche from 2015 to 2017. Dr. Suliman worked for Genentech as a group leader and project team leader in the R&D Portfolio Management and Operations Group from 2010 to 2015, where she oversaw an early development portfolio of more than 30 programs across multiple therapeutic areas. She held various management roles of increasing responsibility at Gilead Sciences, Inc. between 2005 and 2010 and played a significant role in forward-integrating Gilead into new therapeutic areas through M&A. Prior to Gilead, Dr. Suliman was an investment banker with Lehman Brothers and Petkevich & Partners, advising numerous public and private companies on buy- and sell-side transactions. She was named as one of the 2017 Fiercest Women in Life Sciences and as one of the National Diversity Council’s Power 50 in 2021 (Most Influential Women in Business). Dr. Suliman serves as an independent director on the Board of Directors of Ultragenyx Pharmaceutical Inc. (NASDAQ: RARE) and 10x Genomics (NASDAQ: TXG). Dr. Suliman received her M.D. (MB, ChB) from the University of Cape Town Medical School, South Africa, and holds an MBA, with distinction, and M.Phil. degrees from Oxford University, where she was a Rhodes Scholar.
About ReCode Therapeutics
ReCode Therapeutics is an integrated genetic medicines company developing disease-modifying therapeutics using its powerful LNP delivery technology to target organs and tissues beyond the liver. The Company’s pipeline includes lead programs for patients with life-limiting genetic respiratory diseases, including cystic fibrosis and primary ciliary dyskinesia. The Company is leveraging its proprietary LNP platform and nucleic acid technologies and utilizing systemic and direct delivery for mRNA-mediated replacement and gene editing/correction in target cells, including stem cells. For more information, visit www.recodetx.com and follow us on Twitter @ReCodeTx and LinkedIn.
Media Contact:
Will Zasadny
Canale Communications, Inc.
Will.zasadny@canalecomm.com
(619) 961-8848
Investor Contact:
Sarah McCabe
Stern Investor Relations
sarah.mcCabe@sternir.com
IR@recodetx.com
– Alain Baron, M.D., Escient Co-founder and CEO Retires –
SAN DIEGO, Calif., January 5, 2022 — Escient Pharmaceuticals, a clinical-stage company focused on discovering and developing novel, first-in-class therapies to address serious, unserved medical needs, today announced the appointment of Joshua Grass, MBA as Chief Executive Officer effective immediately. Mr. Grass will join the company’s Board of Directors. He succeeds Alain Baron, M.D., who recently retired and will remain a strategic advisor to Escient through the first quarter of 2022.
“I am very excited to be leading Escient during this next phase in its evolution,” said Mr. Grass. “The team has developed very elegant science in characterizing several Mrgprs as potentially important drug targets for large unmet needs. Now, with drug candidates against these targets, the company is poised to translate this science into important medicines for neuroinflammatory and immune activation diseases.”
Prior to joining Escient, Mr. Grass most recently served as CEO of Modis Therapeutics. In 2017, Mr. Grass launched ModisTx with a $30 million Series A financing while working at F- Prime Capital as an Entrepreneur in Residence. ModisTx was acquired by Zogenix in 2019 for $250 million upfront and $400 million in total consideration. Before joining F-Prime, Mr. Grass was a member of the BioMarin senior executive management team. During his 15-year tenure there, he led the acquisition and divestiture of many technologies and products and participated in the global development and launch of multiple products to treat rare genetic diseases. He started his career in biotech as a scientist at Cerus Corporation before holding roles in finance and equity research at boutique investment banks. Mr. Grass earned a Bachelor of Science in Biology from California Polytechnic State University, San Luis Obispo and an MBA in Finance and Entrepreneurship from the William E. Simon School of Business at the University of Rochester.
“Josh’s deep industry experience and impressive track record combined with his scientific and business background and dynamic leadership style make him the right leader for Escient. We are excited to have him at the helm and welcome him to the role.” said Marcus Boehm, Ph.D., Escient’s Co-founder and Chief Scientific Officer.
“On behalf of the Board of Directors and the entire team at Escient, I want to thank Alain for his leadership in building a world class company and team. Escient is well-positioned for the next stage of growth in Mrgpr drug discovery and clinical development,” added Kathleen Sereda Glaub, Chair of the Board of Directors at Escient.
About Escient Pharmaceuticals
Escient Pharmaceuticals is a clinical-stage company focused on discovering and developing novel, first-in-class therapies to address serious, unserved medical needs. The two most advanced programs are focused on new chemical entity antagonists targeting MrgprX4 and MrgprX2 for the treatment of cholestatic/uremic pruritus, and a broad range of mast-cell mediated disorders, respectively. Based in San Diego, Calif., Escient is led by an experienced team of biotechnology entrepreneurs with specific expertise in GPCR drug discovery and development and funded by top-tier life science investors, including The Column Group, 5AM Ventures, Sanofi Ventures, Cowen Healthcare Investors, Redmile Group, Perceptive Advisors, and Osage University Partners.
Visit www.escientpharma.com to learn more.
Contact:
Cory Tromblee
Scient Public Relations
cory@scientpr.com
Svetlana Makhni
Escient Pharmaceuticals, Inc.
info@escientpharma.com
Acquisition will give Aetion customers ability to tap into previously inaccessible, high utility, global health data to conduct transformational healthcare research.
NEW YORK, NY and OTTAWA, ON, January 4, 2022 — Aetion, the leading regulatory-grade real-world evidence (RWE) technology provider, today announced that it has acquired Replica Analytics, a pioneer in using artificial intelligence (AI) for synthetic health data generation.
Replica’s AI technology generates synthetic, privacy-protected copies of real-world data (RWD) that preserve the integrity and utility of source data. Through Replica, Aetion will open up new avenues for better understanding treatment pathways, safety and effectiveness, and improve standards of care.
“Replica enhances Aetion’s technology portfolio to open access to previously inaccessible RWD,” said Dr. Jeremy Rassen, Co-Founder and President of Aetion. “Our shared belief in rooting technology innovation in scientific rigor assures that together Replica and Aetion will accelerate the impact of RWE on improving access to higher quality, more affordable healthcare, globally.”
Replica’s advanced technology expands Aetion’s offerings, including its fit-for-purpose data services, by giving customers more privacy-protected data options when using the Aetion Evidence Platform®—an especially important feature in rare disease and specialty research. Fortune 50 companies already use Replica’s technology to enable fast and effective access to high utility, synthetic versions of health data across their organizations while meeting regulatory obligations and compliance with privacy laws like the Health Insurance Portability and Accountability Act (HIPAA) in the U.S. and the General Data Protection Regulation (GDPR) in Europe. These customers include some of the most prominent life sciences research organizations in the world.
“Despite the tremendous number of valuable RWD sources, the vast majority of the world’s health data is still inaccessible to researchers because of privacy concerns,” said Dr. Khaled El Emam, Replica Analytics’ Co-Founder and CEO, now SVP & General Manager of Replica Analytics at Aetion. “Replica’s mission is to protect that privacy while making the world’s health data responsibly accessible for decision-grade RWE. Becoming part of Aetion and its ecosystem enables us to accelerate and expand that mission by deploying our technology with more data sources and in new applications.”
Aetion’s acquisition of Replica Analytics follows a series of recent high-profile announcements for the company. Most recently, the European Medicines Agency (EMA) selected Aetion to support safety and efficacy research in Europe. Also in 2021, Aetion announced a collaboration with the U.S. Food and Drug Administration (FDA) to generate RWE on inpatient COVID-19 therapies, a partnership with Paris-based AI solutions company Quinten Health to reduce research timelines, and a collaboration with England’s National Institute for Health and Care Excellence (NICE) to explore how RWE can be used to study real-world clinical effectiveness.
About Aetion
Aetion is a healthcare analytics company that delivers real-world evidence for the manufacturers, purchasers, and regulators of medical treatments and technologies. The Aetion Evidence Platform® analyzes data from the real world to produce transparent, rapid, and scientifically validated answers on safety, effectiveness, and value. Founded by Harvard Medical School faculty members with decades of experience in epidemiology and health outcomes research, Aetion informs healthcare’s most critical decisions—what works best, for whom, and when—to guide product development, commercialization, and payment innovation.
Aetion is based in New York City and backed by investors including Warburg Pincus, New Enterprise Associates (NEA), Flare Capital Partners, Greenspring Associates, Lakestar, B Capital, Foresite Capital, Town Hall Ventures, McKesson Ventures, Sanofi Ventures, EDBI, Johnson & Johnson Innovation—JJDC, Inc., UCB, Amgen Ventures, and Horizon Health Services, Inc. Learn more at aetion.com and follow us at @aetioninc.
About Replica Analytics
Replica Analytics is a developer of unique technologies for generating privacy protective synthetic data that maintains the statistical properties of real data. The Replica Synthesis software provides a full suite of synthetic data generation and evaluation capabilities that can solve multiple grand challenges facing the life sciences industry, and health research in general. Synthetic data enables rapid innovation by accelerating the development of AI models and accelerating clinical studies through data simulations. For more information, visit: https://replica-analytics.com/
Aetion
+1-844-823-8466
US: +1-646-513-2200
Europe: +33-983-887-249
New York
Headquarters 5 Penn Plaza 7th Floor
New York, NY 10001
Boston
Science & Technology Office
50 Congress Street
Suite 1025
Boston, MA 02109
Los Angeles
Science & Technology Office
840 Apollo Street
Suite 100
El Segundo, CA 90245
Barcelona
European Office
Can Rabia 3-5, Planta 4
08017 Barcelona
Spain
SAN CARLOS, Calif., December 17, 2021 – Glycomine, Inc., a biotechnology company focused on developing new therapies for orphan diseases, today announced that the company has received U.S. Food and Drug Administration (FDA) clearance of an Investigational New Drug (IND) application for GLM101 for the treatment of PMM2-CDG and has initiated dosing healthy volunteers in a Phase 1 clinical study. GLM101 is a mannose-1-phosphate replacement therapy in development to treat phosphomannomutase 2-congenital disorder of glycosylation (PMM2-CDG), previously known as CDG Type Ia. PMM2-CDG is the most prevalent congenital disease of glycosylation but has no FDA-approved treatments.
“The initiation of this clinical study is an important milestone for Glycomine and for PMM2-CDG patients and their families,” said Peter McWilliams, Ph.D., CEO of Glycomine. “We have demonstrated in preclinical studies that GLM101 can restore the glycosylation pathway that is disrupted in PMM2-CDG, and we look forward to advancing our clinical studies to confirm the potential of GLM101 as an important disease-modifying therapy for PMM2-CDG patients.”
“PMM2-CDG is a serious rare disease with no therapeutic options and represents a critical unmet medical need,” said Horacio Plotkin, M.D., FAAP, Chief Medical Officer of Glycomine. “Glycomine has developed a proprietary approach to delivering mannose-1-phosphate intracellularly to overcome the deficiency that leads to PMM2-CDG. The open-label Phase 1 study will evaluate safety and tolerability in healthy volunteers. With successful completion, we look forward to opening enrollment for patients, which is planned for the second half of 2022.”
About GLM101, a Potential Treatment for PMM2-CDG (CDG-Ia)
Glycomine’s GLM101 is a mannose-1-phosphate replacement therapy in development to treat PMM2-CDG. PMM2-CDG is a severe multisystem disorder with symptoms such as coagulopathies, endocrinopathies, hypotonia, stroke-like episodes, as well as immune and nervous system disfunctions, and resulting mortality of 20% in the early years of life. PMM2-CDG is caused by genetic mutations that lead to a deficiency of the enzyme phosphomannomutase 2 (encoded by the PMM2 gene). PMM2 converts mannose-6-phosphate to mannose-1-phosphate, which is an essential sugar molecule in the N-glycosylation pathway and is crucially important for proper glycoprotein structure and function. GLM101 is designed to deliver mannose-1-phosphate directly into cells and thereby bypass the PMM2 enzyme deficiency and address all disease-causing PMM2 mutations to restore pathway function. GLM101 has received Orphan Drug Designation in the U.S. and Europe and Rare Pediatric Disease Designation in the U.S.
About Glycomine, Inc.
Glycomine is developing orphan drugs for serious rare disorders of metabolism and protein misfolding for which no other therapeutic options exist. The company's approach is to use replacement therapies – substrates, enzymes, or proteins – and to target those molecules to clinically relevant cellular compartments. The company is based in San Carlos, California and supported by leading international life sciences investors.
Corporate Contact: Peter McWilliams, Ph.D., info@glycomine.com
Media Contact: Jessica Yingling, Ph.D., Little Dog Communications Inc., jessica@litldog.com, +1.858.344.8091
- Study of lead candidate to provide important information on safety, pharmacokinetics and pharmacodynamics to inform further clinical development in chronic inflammatory diseases
- Highlights continued progress advancing a differentiated portfolio of novel NLRP3 inflammasome inhibitors to treat both peripheral- and neuro- inflammatory diseases
BOSTON, SEATTLE and CAMBRIDGE, UK – November 4, 2021 – NodThera, a clinical-stage biotechnology company developing a new class of medicines that inhibit the NLRP3 inflammasome to treat chronic inflammation, today announced that the first healthy volunteers have been dosed in a Phase 1 clinical trial of its lead investigational candidate, NT-0796.
NT-0796 is a small molecule NLRP3 inflammasome inhibitor with differentiated novel chemistry that provides unprecedented potency and potential for prolonged pharmacodynamic (PD) effect, with the ability to cross the blood brain barrier in preclinical species. NT-0796 selectively inhibits NLRP3, the upstream regulator of the body’s inflammation response, to reduce levels of both IL-1β and IL-18 – pro-inflammatory cytokines known to play a role in chronic inflammation underlying a wide range of chronic diseases. Pharmacokinetic (PK) and PD data from an ex vivo IL-1β/IL-18 stimulation assay and cerebrospinal fluid (CSF) sampling in the Phase 1 study will inform further clinical development.
“The recent convergence of key insights into innate immunity, IL-1β/IL-18 and the NLRP3 inflammasome have revolutionized our understanding of chronic disease,” said Adam Keeney, Ph.D., CEO of NodThera. “As one of the first companies to recognize the importance of NLRP3 in the inflammation cascade, we look forward to gathering important human data from the Phase 1 clinical study of our lead candidate NT-0796 so we can accelerate innovation for patients with limited treatment options.”
The primary objective of this study is to assess the safety and tolerability of NT-0796, while secondary objectives include assessment of PK and PD (ability to lower IL-1 and IL-18 levels) and CSF sampling to assess NLRP3 target engagement and compound exposure after single and multiple ascending doses.
“NT-0796 leverages novel chemistry that is unlike any other NLRP3 inflammasome inhibitor in the field. It is designed to deliver key advantages in PK and PD, with the potential to cross the blood brain barrier,” said Donald Johns, M.D., Chief Medical Officer of NodThera. “The NLRP3 inflammasome is a key driver of diseases that span different parts of the body, from common ailments such as osteoarthritis, to cardiovascular disease, Alzheimer’s Disease, cancer, and beyond. Unlocking this treatment potential provides the opportunity to impact many patients whose quality of life is negatively affected by chronic inflammatory disease.”
About NodThera
NodThera is a clinical-stage biotechnology company developing a new class of potent and selective NLRP3 inflammasome inhibitors for the treatment of diseases driven by chronic inflammation. Led by an experienced management team, NodThera is leveraging new insights into inflammasome biology and chemistry to build a portfolio of highly differentiated small molecule NLRP3 inflammasome inhibitors. The company was founded in 2016 and financed by 5AM Ventures, Cowen Healthcare Investments, Epidarex Capital, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera is headquartered in Lexington, MA, with additional locations in Cambridge, UK and Seattle, WA. Learn more at www.nodthera.com or follow us on LinkedIn.
Media Contact
Darby Pearson
Verge Scientific Communications
dpearson@vergescientific.com
Brings more than 15 years of CNS drug discovery and development experience
Copenhagen, Denmark and Boston, US, 1st November 2021 – Muna Therapeutics (“Muna”), pioneering the development of novel, first-in-class small molecule therapeutics for neurodegenerative diseases, today announces the appointment of Dr. Niels Plath as Chief Scientific Officer.
Dr. Plath is a molecular biologist and neuroscientist with more than 15 years of experience in academia and biopharma. He was previously the acting Head of Global Research at Lundbeck, leading drug discovery and development for neurologic and psychiatric diseases. Prior to this role, Dr. Plath was Vice President for Neuroscience, leading teams that brought several drug candidates into clinical development, including alpha-synuclein and tau targeting antibodies, idalopirdine and Nalmefene.
Rita Balice-Gordon, Chief Executive Officer of Muna Therapeutics, said: “We are delighted to welcome Niels as Muna’s Chief Scientific Officer. He shares our commitment to discover and develop disease modifying therapeutics to address the staggering unmet need experienced by patients with neurodegenerative disorders around the world.”
Henrijette Richter, Managing Partner at Sofinnova Partners, said: “Niels has broad biopharma R&D experience that will be invaluable as Muna continues to progress its innovative pipeline of potential disease-modifying medicines for neurodegenerative diseases. We are very pleased to be supporting Muna’s world-class global team.”
Dr. Niels Plath, Chief Scientific Officer of Muna Therapeutics, said: “I’m excited to be joining Muna Therapeutics to further develop its promising, cutting-edge pipeline of neurodegenerative disease therapeutics. I’m looking forward to working with this stellar team to advance promising treatments for patients to significantly improve their quality of life.”
Dr. Plath obtained a PhD in Neuroscience from the Free University of Berlin, Germany, focusing on the role of immediate early genes in neuronal plasticity, learning and memory. Following a postdoctoral fellowship supported by the Human Frontier Science Program, he joined biopharma in 2005 to pursue research and development of treatments for patients with CNS disorders. Dr. Plath has authored many scientific publications in peer-reviewed journals, is a guest lecturer in Neuroscience at the University of Copenhagen and has given numerous conference talks at meetings around the world.
ENDS
For more information please contact:
Muna Therapeutics
Rita Balice-Gordon, CEO
Email: balicegordon@munatherapeutics.com
Optimum Strategic Communications
Mary Clark, Stella Lempidaki, Zoe Bolt
Tel: +44 (0) 20 3922 1906
Email: muna@optimumcomms.com
About Muna Therapeutics
Muna Therapeutics is a private biopharmaceutical company founded in 2020 and based in Copenhagen, Denmark and Leuven, Belgium. Muna discovers and develops therapies that slow or stop devastating neurodegenerative diseases including Alzheimer’s, Frontotemporal Dementia and Parkinson’s. These disorders impact memory, movement, language, behavior and personality resulting in disability and death of millions of patients around the globe. We focus our groundbreaking science on identifying new medicines to preserve cognition and other brain functions and enhance resilience to neurodegenerative diseases. Our name reflects this focus: Muna means ‘to remember’ in Old Norse. www.munatherapeutics.com

NEW YORK--(BUSINESS WIRE)--Ovid Therapeutics Inc. (Ovid), a privately held biopharmaceutical company focused on developing therapies for rare and orphan diseases of the brain, today announced that Bart Friedman has joined the Company’s Board of Directors. Mr. Friedman is a senior partner at the Wall Street law firm Cahill Gordon & Reindel LLP and is Chairman of its Business Development Committee.
“We are delighted that Bart is joining our Board. Our goal is to build a world-leading neurology company with a Board of unparalleled and diverse capabilities. Bart’s broad experience, exceptional reputation, deep knowledge, and intellect are exactly what we require to achieve this goal,” said Dr. Jeremy Levin, Chief Executive Officer and Chairman of Ovid. “It is a privilege to have the opportunity to work with him as we seek to bring medicines to patients and help the families of those with these significant disorders.”
At Cahill Gordon & Reindel, Mr. Friedman advises leading financial institutions and global corporations, boards of directors, audit committees, and officers and directors of publicly held companies in significant corporate and securities matters, corporate compliance, and enforcement challenges. Mr. Friedman chairs the Board of Directors of the Sanford C. Bernstein Mutual Funds, is the Lead Independent Director of Allied World Assurance Holdings, serves on the Board of Trustees of The Brookings Institution and chairs its Audit Committee, and serves on the Board of Lincoln Center for the Performing Arts and chairs its Audit Committee. Mr. Friedman also serves on the Membership Committee of the Council on Foreign Relations. Earlier in his career, Mr. Friedman worked at the Securities and Exchange Commission as Special Counsel and later as Assistant Director. He earned his J.D. at Harvard Law School and was on the research faculty of Harvard Business School during and following his graduation from Harvard Law School and prior to joining Cahill.
Mr. Friedman commented, “I have been impressed with Ovid’s mission, strategy, and progress as it becomes a pre-eminent global, commercial company focused on orphan diseases of the brain. This is an important area of medical need, and Ovid is well-positioned to play a leading role. I am honored to serve on Ovid’s Board.”
About Ovid Therapeutics Inc.
Ovid Therapeutics Inc. is a privately held, New York-based, biopharmaceutical company committed to transforming the lives of patients with rare and orphan diseases of the brain. Ovid focuses on patients and their unmet medical needs. Using the significant operational, product development, and business development experience of its management team, Ovid aims to become a leading neurology company, with multiple products and a rich pipeline, coupled with compelling research and development. Ovid recently completed a substantially oversubscribed $75 Million Series B financing led by Fidelity Management and Research Company and including Cowen Private Investments, Sanofi-Genzyme BioVentures, Tekla Capital Management, Sphera Global Healthcare Fund, Jennison Associates (on behalf of certain clients), Redmile Group, DoubleLine Equity Healthcare fund, and Cormorant Asset Management, as well as additional blue chip mutual funds and leading life sciences investors.
For more information on Ovid, please visit http://www.ovidrx.com.
Contacts
Burns McClellan, on behalf of Ovid Therapeutics
Justin Jackson, 212-213-0006, ext. 327
jjackson@burnsmc.com
NEW YORK--(BUSINESS WIRE)--Click Therapeutics, Inc. (“Click”), a leader in Digital Therapeutics™ as prescription medical treatments, today announced that it has closed a $52 million upsized Series B financing. The financing round was co-led by new investors H.I.G. BioHealth Partners (“H.I.G. BioHealth”) and Accelmed Partners II (“Accelmed”), with participation from Health Catalyst Capital, Revelation Partners, and a top biotechnology hedge fund, and from existing investors Sanofi Ventures, K2 HealthVentures, Hikma Ventures, and Ridgetop Health. Proceeds from the Series B financing will be used to accelerate the development and commercialization of Click’s internal prescription digital therapeutic pipeline and advance Click’s platform capabilities.
Click’s pipeline of innovative digital therapeutics spans multiple therapeutic areas, from psychiatry and chronic pain to cardiometabolic and autoimmune disorders. In addition to its internal pipeline programs, Click has entered into landmark collaboration agreements with Otsuka to develop and commercialize a prescription digital therapeutic for treatment of Major Depressive Disorder (MDD), and with Boehringer Ingelheim to develop and commercialize a prescription-based digital therapeutic to aid in the treatment of schizophrenia.
“Closing our Series B is an important milestone for Click that will allow us to further scale our proprietary Click Neurobehavioral Intervention (CNI) Platform and fund the development of new and innovative digital therapeutics for patients in need,” said David Benshoof Klein, Co-Founder and CEO of Click. “We are excited to welcome our new investors and deepen our relationships with existing investors, all of whom share our vision of a new healthcare landscape in which prescription digital therapeutics play a prominent role alongside traditional pharmacological treatments.”
In conjunction with the Series B financing, Click’s Board of Directors will expand to include Alex Zisson, Managing Director at H.I.G. BioHealth, and Dr. Uri Geiger, Founder and Managing Partner of Accelmed.
“Digital therapeutics hold the promise to change the very paradigm of healthcare,” said Mr. Zisson. “Click has one of the very best platforms in this field, and we are excited to help the company improve patient outcomes and potentially lower healthcare costs.”
“Click is a prime example of the tremendous and growing potential for digital health software applications to enhance the lives of patients suffering from a variety of medical conditions,” commented Dr. Geiger. “Click is the ideal partner for Accelmed due to our shared belief that leveraging technology in healthcare improves patient outcomes and increases efficiencies throughout the healthcare system. We are excited to offer our financial and operational expertise and to work closely with David and the entire Click executive team to support the growth of Click’s innovative platform.”
About Click Therapeutics
Click Therapeutics, Inc. develops and commercializes software as prescription medical treatments for patients with unmet medical needs. Through cognitive and neurobehavioral mechanisms, Click’s Digital Therapeutics™ enable change within individuals, and are designed to be used independently or in conjunction with biomedical treatments. The Clickometrics® adaptive data science platform continuously personalizes user experience to optimize engagement and outcomes. Following a groundbreaking clinical trial, Click’s industry-leading smoking cessation program is available nationwide through a wide variety of payers, providers, and employers. Click’s lead prescription program has recently entered a pivotal, fully remote, randomized, controlled trial on the Verily platform for the treatment of Major Depressive Disorder in up to 540 adults. For more information on Click, visit ClickTherapeutics.com.
About H.I.G. BioHealth Partners
H.I.G. BioHealth Partners is the dedicated life-science investment affiliate of H.I.G. Capital. H.I.G. BioHealth Partners invests in a broad range of healthcare opportunities across sectors and stages, principally in companies developing therapeutic drugs, medical devices, and diagnostics for significant unmet medical needs. With approximately $400 million in committed capital, H.I.G. BioHealth Partners invests $5 million to $40 million per company over the life of an investment. For more information, please refer to the H.I.G. BioHealth Partners website at www.higbio.com.
About Accelmed Partners
Accelmed is a U.S.-based private equity firm focused on acquiring and investing in U.S. commercial stage, lower middle market HealthTech companies. Since 2009, Accelmed has deployed over $400 million into companies spanning medical devices, diagnostics, digital health and technology-enabled healthcare services. Accelmed seeks to accelerate value and scale innovation across the HealthTech field by bringing to bear the team’s industry experience, operational and financial expertise, and strong global relationships. For more information, please visit www.accelmed.com.
Contacts
Company Contact
Daniel Busby
VP, Investor Relations & Strategic Finance
dbusby@clicktherapeutics.com
Media Contact
Karen Sharma
ksharma@macbiocom.com
781-235-3060
The agency will use Aetion’s scientifically validated software platform to develop a system of studies that enables rapid evidence generation for COVID-19 and future pandemics
NEW YORK, NY, October 21, 2021 — Today, Aetion announces that it is expanding its relationship with the U.S. Food and Drug Administration (FDA) to use real-world evidence (RWE) to study COVID-19 interventions and advance regulatory science and innovation. FDA and Aetion will use Aetion Evidence Platform® (AEP), validated software that enables efficient, transparent, and reliable RWE research, to develop a framework and system of studies for the rapid assessment of COVID-19 inpatient medical products.
This project is designed to demonstrate how using a platform-based approach furthers regulatory learnings on the use of RWE to inform decision-making. The work will also provide a scalable infrastructure for the rapid development and evaluation of COVID-19 therapies, which can be applied for future public health emergencies. This contract will support the agency’s broader digital transformation efforts, which include its Technology and Data Modernization Action Plans as well as its recently announced Office of Digital Transformation.
“COVID-19 has created an urgent need to develop and apply innovative methods to assess novel interventions,” said Dr. Jeremy Rassen, co-founder and president of Aetion. “As FDA continues to advance its digital capabilities, Aetion is proud to partner with the agency in developing the rigorous scientific processes and RWE generation tools needed to quickly respond to future public health challenges.”
The aim of this project is to further data familiarity and protocol standards to support real-world data (RWD) analyses among the broader research community. FDA will work with Aetion to define and prioritize key research questions; identify fit-for-purpose data sources; develop appropriate, validated, and applicable measurement algorithms to capture key exposures, subgroups, confounding variables, and outcomes; design template epidemiological studies applicable to a range of treatments; implement studies and generate transparent reporting using AEP; and build and disseminate knowledge via peer-reviewed publications and other avenues.
Aetion and FDA will build on learnings from the research collaboration announced in May 2020, in which FDA and Aetion explored the utility of RWD to advance the understanding of and response to COVID-19. Since launching the collaboration, FDA and Aetion have developed mechanisms to assess data fitness for use; identified methodological good practices on working with RWD for COVID-19; and built the foundation for rapid-cycle analytics to address critical and emergent public health questions.
About Aetion
Aetion is a health care analytics company that delivers real-world evidence for the manufacturers, purchasers, and regulators of medical treatments and technologies. The Aetion Evidence Platform® analyzes data from the real world to produce transparent, rapid, and scientifically validated answers on safety, effectiveness, and value. Founded by Harvard Medical School faculty members with decades of experience in epidemiology and health outcomes research, Aetion informs health care’s most critical decisions— what works best, for whom, and when—to guide product development, commercialization, and payment innovation.
Aetion is based in New York City and backed by investors including New Enterprise Associates (NEA), Warburg Pincus, Flare Capital Partners, Greenspring Associates, Lakestar, B Capital, Foresite Capital, Town Hall Ventures, McKesson Ventures, Sanofi Ventures, EDBI, Johnson & Johnson Innovation—JJDC, Inc., UCB, Amgen Ventures, and Horizon Health Services, Inc. Learn more at aetion.com and follow us at @aetioninc.
+1-844-823-8466
US: +1-646-513-2200
Europe: +33-983-887-249
New York
Headquarters
5 Penn Plaza 7th Floor
New York, NY 10001
Boston
Science & Technology Office
50 Congress Street
Suite 1025
Boston, MA 02109
Los Angeles
Science & Technology Office
840 Apollo Street Suite 100
El Segundo, CA 90245
Barcelona
European Office
Can Rabia 3-5, Planta 4
08017 Barcelona
Spain
- Financing co-led by Pfizer Ventures and EcoR1 Capital with participation from syndicate of world-class life sciences investors
- Company to advance mRNA and gene correction therapies into the clinic for cystic fibrosis and primary ciliary dyskinesia
- ReCode’s proprietary non-viral lipid nanoparticle (LNP) delivery platform to generate a deep pipeline of therapies that target the lung, liver and other extra-hepatic tissues
- Company to expand internal manufacturing capabilities to support research and clinical programs
Menlo Park, Calif. and Dallas, Texas – October 21, 2021 – ReCode Therapeutics (the “Company”), a biopharmaceutical company pioneering disease-modifying genetic medicines using its proprietary LNP delivery platform, today announced the closing of an $80 million Series B financing round co-led by Pfizer Ventures and EcoR1 Capital. New investors include Sanofi Ventures, funds managed by Tekla Capital Management LLC, Superstring Capital and NS Investment. Existing investors who participated included OrbiMed, Vida Ventures, MPM Capital, Colt Ventures, Hunt Technology Ventures, L.P., and Osage University Partners (OUP). The proceeds from the Series B financing will be used to drive ReCode’s lead programs in primary ciliary dyskinesia (PCD) and cystic fibrosis (CF) into human clinical studies, expand the pipeline of treatments for patients with life-limiting genetic respiratory diseases, advance its LNP platform for organ-specific delivery of RNA and gene correction therapies and increase internal manufacturing capabilities.
“ReCode is working to unleash the power of genetic medicine by delivering therapies with our novel LNP platform, which has the potential to reach across a broad spectrum of diseases involving multiple organs and tissues,” said David Lockhart, Ph.D., CEO and president, ReCode Therapeutics. “The significant capital secured from such a respected group of investors, known for backing innovative biotechnology companies, enables us to accelerate delivery of impactful medicines to thousands of patients with genetic respiratory diseases in need of options, including those with CF and PCD.”
In connection with the closing of the financing, Rana Al-Hallaq, Ph.D., a partner at Pfizer Ventures and executive director for Pfizer Worldwide Business Development, has joined the ReCode board of directors. “Through this investment, we are excited to support ReCode in its development of these novel LNPs, which we believe, if successful, may significantly expand the potential of genetic medicine across therapeutic areas,” Al-Hallaq said.
Oleg Nodelman, founder and portfolio manager of EcoR1 Capital also joined ReCode’s Board of Directors in connection with the financing. “We are excited to co-lead ReCode’s Series B financing and to support the company as they advance their unique technology that enables the delivery of novel genetic medicines to target organs, tissues and cells. ReCode’s platform has the potential to unlock vast capabilities unaddressable by first-generation mRNA and gene editing programs and enable development of therapeutics for patients with diseases that have historically been untreatable.”
ReCode’s lead programs are focused on the genetic respiratory diseases PCD and CF. Recent preclinical data from the Company’s RNA-based CF program showed that its LNPs can deliver CFTR mRNA that restores cystic fibrosis transmembrane conductance regulator (CFTR) function in the CF patient-derived hBE cell model. Preclinical data from the Company’s inhaled mRNA-based program for the treatment of PCD demonstrated that its LNP formulations successfully delivered DNAI1 mRNA to target airway epithelial cells in hBEs, mice and NHPs, and that robust ciliary activity was restored in treated DNAI1- deficient hBE cells.
About ReCode Therapeutics
ReCode Therapeutics is an integrated genetic medicines company developing disease-modifying therapeutics using its powerful LNP delivery technology to target organs and tissues beyond the liver. The Company’s pipeline includes lead programs for patients with life-limiting genetic respiratory diseases, including cystic fibrosis and primary ciliary dyskinesia. The Company is leveraging its proprietary LNP platform and nucleic acid technologies and utilizing systemic and direct delivery for mRNA-mediated replacement and gene editing/correction in target cells, including stem cells. For more information, visit www.recodetx.com and follow us on Twitter @ReCodeTx and LinkedIn.
Media Contact:
Will Zasadny
Canale Communications, Inc.
will.zasadny@canalecomm.com
(619) 961-8848
Investor Contact:
Sarah McCabe
Stern Investor Relations
sarah.mccabe@sternir.com
IR@recodetx.com
BOSTON, MA—October 12, 2021—i2O Therapeutics, a developer of novel peptide and antibody based oral biologic products based on proprietary ionic liquid platform, announces the appointment of Kurt C. Graves as Executive Chairman. Mr. Graves brings 30 years of business leadership experience in leading global pharmaceutical and biotech companies.
“Kurt’s business acumen and expertise in setting vision, culture, and growth strategies across early, mid and late-stage bio-pharma companies will be invaluable at this pivotal time of corporate growth at i2O Therapeutics,” said Ravi Srinivasan, PhD, CEO of i2O Therapeutics. “Kurt has successfully led the development, launch, and build-out of multiple blockbuster brands and multi-billion dollar franchises, and his deep background in over 15 therapeutic areas, crafting major financings and partnerships, and working with peptides and biologics makes him the ideal selection to lead our Board.”
Over the last 10 years Mr. Graves has provided leadership to several innovative biotech companies including as Chairman, President and CEO of Intarcia Therapeutics, former Chairman of Radius Health, and as a Board member at Achillion Pharmaceuticals until it was acquired by Alexion, and at Seres Health. He was also E&Y’s New England Entrepreneur of the Year in 2015. Previously, Mr. Graves served as EVP, Head of Corporate & Business Development, Head of Strategic Drug Development and Program Management & Commercial at Vertex Pharmaceuticals. Prior to that, he was at Novartis Pharmaceuticals for nearly 10 years, most recently as the Global Head of the General Medicines Business & the first Global Chief Marketing Officer for the Pharmaceuticals division. Earlier in his career, at Merck and Astra-Merck he led the GI Business Unit responsible for Prilosec , Nexium , and Prilosec OTC .
“i20 Therapeutics has developed a transformational new platform for enabling the full potential of oral biologics including peptides, antibodies and nucleic acids. The company is focused on creating novel oral biologics that are also tunable and well-differentiated therapies that can provide superior clinical efficacy and safety benefits to patients. Our vision and scope of potential therapeutic impact is very exciting and directly aligns with my passion to help develop novel platform technologies and therapeutics that meaningfully enhance lives and real-world outcomes,” said Graves. “I look forward to working with the management team and Board and forming key external partnerships to help scale our platform, strengthen the organization, and advance a new generation of tunable oral biologics starting with programs in serious immune-mediated inflammatory diseases and metabolic therapeutic areas.
Mr. Graves received a B. S. in Biology from Hillsdale College and has attended educational and leadership development programs at Harvard University, University of Michigan, and the Wharton School of Management.
About i2O Therapeutics
i2O Therapeutics is the leader in exploiting the versatile properties of Ionic Liquids (ILs) for therapeutic development. i2O’s wholly owned pipeline consists of first-in-class and best-in-class transformative oral biologics with improved clinical benefits and a superior safety profile over current standard of care. With a focus on metabolic diseases, inflammatory diseases and other indications, i2O is delivering on the promise of oral biologics—considered the “holy grail biologic opportunity” by the pharmaceutical industry. Visit us at www.i2obio.com.
- Science 37 to debut on Nasdaq as a publicly traded company under ticker symbol “SNCE”
- Business combination will provide Science 37 with approximately $235 million in cash proceeds to support continued growth
- Science 37 to fuel its mission to enable universal access to clinical research which has proven to accelerate patient enrollment, minimize patient burden, and include underserved patient populations
- Investments targeted to enhance the Science 37 Operating System to decentralize clinical trial execution and enable more agile clinical trials
LOS ANGELES and NEW YORK, October 7, 2021 – Science 37, Inc., the Operating System for today’s agile clinical trials, announced today that it has completed its previously announced business combination with LifeSci Acquisition II Corp. (NASDAQ: LSAQ) (“LifeSci”), a blank check company targeting the biopharma, medical technology, digital health and healthcare services sectors. Shares of common stock of the combined company, named Science 37
Holdings, Inc. (“Science 37” or the "Company") will begin trading on the Nasdaq Global Market under the new ticker symbol "SNCE” today, October 7, 2021.
LifeSci shareholders approved the transaction at a special meeting on October 4, 2021. As a result of the transaction, Science 37 has received approximately $235 million total cash, net of fees and expenses paid in connection with the closing of the business combination, including the proceeds from the private placement completed in connection with the transaction. Science 37 intends to use the transaction proceeds to fund its decentralized trial technology platform, extend into new adjacencies, and power the next generation in clinical research. David Coman, Chief Executive Officer of Science 37, Inc., and Science 37 Inc.’s current executive team will continue to lead the combined company.
“The Science 37 Operating System has been proven to materially accelerate patient enrollment, provide meaningfully higher patient retention, significantly reduce patient burden and enable participation from underserved patient populations. This is all made possible through our full-stack, end-to-end technology platform and supported by specialized patient communities, telemedicine investigators, mobile nurses, and remote coordinators,” said Mr. Coman. “The additional capital from this transaction will help us deliver on our vision to be the category-defining operating system that powers every clinical trial as the industry shifts to more agile trial designs.”
Andrew McDonald, Ph.D., Chief Executive Officer of LifeSci, said, “Science 37’s extraordinary differentiation based on its technology platform and specialized networks represents the future of clinical trial operations. As a leader and innovator in agile clinical trials, Science 37 has the opportunity to transform the industry and impact positive change in millions of people's lives. We are proud to partner with David and his talented team as Science 37 begins its next chapter as a public company.”
Following the business combination, David Coman will serve on Science 37’s Board of Directors alongside Rob Faulkner, Managing Director at Redmile Group, as Chairman; John W. Hubbard, Ph.D., former Chief Executive Officer at Bioclinica and former Senior Vice President and Worldwide Head of Development Operations at Pfizer; Bhooshi de Silva, Senior Vice President, Global Head of Corporate Development, Strategy and Ventures, at PPD; Adam Goulburn, Ph.D., Partner at Lux Capital; Neil Tiwari, Partner of Private Healthcare Ventures at Magnetar Capital; and Emily Rollins, former Partner of Deloitte & Touche.
Advisors
Cowen and Perella Weinberg Partners LP served as financial advisors and Latham & Watkins LLP and DLA Piper LLP (US) served as legal advisors to Science 37. Cowen acted as sole placement agent to LifeSci Acquisition II Corp. in connection with the private placement. LifeSci Capital LLC acted as lead book-running manager to LifeSci Acquisition II Corp. in connection with its initial public offering in November 2020. Loeb & Loeb LLP served as legal advisor to LifeSci Acquisition II Corp. Cowen, William Blair, Baird, and Lake Street Capital Markets acted as capital markets advisors to Science 37.
About Science 37
Science 37's mission is to enable universal access to clinical research—making it easier for patients and providers to participate from nearly anywhere and helping to accelerate the development of treatments that impact patient lives. As a pioneer of decentralized clinical trials, the Science 37 Clinical Trial Operating System (OS) supports today’s more
agile clinical research designs with its full stack, end-to-end technology platform and specialized networks of patient communities, telemedicine investigators, mobile nurses, remote coordinators and connected devices. Configurable to enable any study type, the Science 37 OS can enable up to 15x faster enrollment, 28% better retention and 3x more diverse patient population with industry-leading workflow orchestration, evidence generation and data harmonization. For more information, visit https://www.science37.com.
About LifeSci Acquisition II Corp.
LifeSci Acquisition II Corp. (Nasdaq: LSAQ) is a blank check company formed for the purpose of entering into a merger, share exchange, asset acquisition, stock purchase, recapitalization, reorganization, or other similar business combination with one or more businesses or entities, pursuing targets that are focused on healthcare innovation in North America or Europe. For more information visit: https://lifesciacquisition.com/spac-2/.
Forward-Looking Statements
This press release contains certain forward-looking statements within the meaning of the federal securities laws, including statements regarding the benefits of the transaction between Science 37, Inc. and LifeSci, the services offered by Science 37 and the markets in which it operates, and Science 37’s projected future results. These forward- looking statements generally are identified by the words “believe,” “project,” “expect,” “anticipate,” “estimate,” “intend,” “strategy,” “future,” “opportunity,” “plan,” “may,” “should,” “will,” “would,” “will be,” “will continue,” “will likely result” and similar expressions. Forward-looking statements are predictions, projections and other statements about future events that are based on current expectations and assumptions and, as a result, are subject to risks and uncertainties. Many factors could cause actual future events to differ materially from the forward-looking statements in this press release, including but not limited to: (i) the ability to maintain the listing of Science 37’s securities on Nasdaq, (ii) volatility in the price of Science 37’s securities due to a variety of factors, including changes in the competitive and highly regulated industries in which Science 37 plans to operate, variations in performance across competitors, changes in laws and regulations affecting Science 37’s business and changes in its capital structure, (iii) the ability to implement business plans, forecasts, and other expectations, and to identify and realize additional opportunities, (iv) the risk that Science 37 may never achieve or sustain profitability, (v) the risk that Science 37 will need to raise additional capital to execute its business plan, which may not be available on acceptable terms or at all; and (vi) the potential adverse effects of the ongoing global COVID-19 pandemic. The foregoing list of factors is not exhaustive.
You should carefully consider the foregoing factors and the other risks and uncertainties described in the “Risk Factors” section of LifeSci’s definitive proxy statement/prospectus and registration statement on Form S-4 filed with the U.S. Securities and Exchange Commission (the “SEC”) and other documents filed by Science 37 from time to time with the SEC. These filings identify and address other important risks and uncertainties that could cause actual events and results to differ materially from those contained in the forward-looking statements. Forward-looking statements speak only as of the date they are made. Readers are cautioned not to put undue reliance on forward-looking statements, and Science 37 and LifeSci assume no obligation and do not intend to update or revise these forward- looking statements, whether as a result of new information, future events, or otherwise. Neither Science 37 nor LifeSci gives any assurance that either Science 37 or LifeSci will achieve their expectations.
Contacts For Media:
Margie Kooman
margie.kooman@science37.com
Nina Gill
Science37@10fold.com
For Investors:
Caroline Paul Gilmartin Group
investors@science37.com
- Completed enrolment of 150 pregnant women in a Phase 2 study in South Africa and dosing started in an additional 50 pregnant women in Uganda, as part of same study
- Phase 1 booster study started in the United Kingdom
- Regulatory approval obtained from Danish Medicines Agency and UK MHRA for the initiation of a Phase 2 study in pregnant women, in Denmark and United Kingdom
- Vaccine Expert and Biotech Entrepreneur Gerd Zettlmeissl appointed new Chairman of the Board of DirectorsLeading vaccine industry veterans join Scientific Advisory Board
Copenhagen, Denmark, 7 October 2021 – MinervaX, a privately held Danish biotechnology company developing a novel vaccine against Group B Streptococcus (GBS), today announces clinical progress on its maternal GBS vaccine as well as multiple additions to its leadership teams.
MinervaX is developing a maternal vaccine for the prevention of adverse pregnancy outcomes and life- threating infections caused by Group B streptococcus (GBS). GBS is responsible for nearly half of all life- threatening infections in newborns during the first 3 months of life as well as a portion of late-term abortions, premature deliveries, or stillbirths during pregnancy. Current preventative strategy is insufficient, and great medical needs exist, which may be addressed by a maternal GBS vaccine. MinervaX’s maternal GBS vaccine is based on adjuvanted proteins antigens covering close to 100% of clinical GBS isolates.
Clinical Update:
MinervaX announces today completed enrolment of 150 pregnant women in a Phase 2 study in South Africa, sponsored by the European Developing Countries Trial Partnership. Dosing of an additional 50 pregnant women in the same study has started in Uganda. The study is listed on clinicaltrials.gov under NCT04596878.
MinervaX has also started dosing healthy adult women previously receiving the Company’s GBS vaccine in a Phase 1 booster study in the United Kingdom. The study is listed on clinicaltrials.gov under NCT05005247.
Finally, MinervaX has received regulatory approval from the Danish Medicines Agency and UK MHRA to initiate a Phase 2 study in 270 pregnant women in Denmark and the United Kingdom.
Corporate Update:
Gerd Zettlmeissl joins MinervaX as new Chairman of the Board of Directors. Gerd brings a wealth of experience from the vaccine and broader biotech industry, including several successful biotech exits.
MinervaX announces the addition of Ralf Clemens, Jean-Paul Prieels, Peter Patriarca, Jean Smal, Clement
Lewin, and Inca Kusters as new members of its Scientific Advisory Board (SAB).
MinervaX ApS, Ole Maaløes Vej 3, DK-2200 Copenhagen N, Denmark VAT no. DK32673287
Commenting on the announcement, Per Fischer, Chief Executive Officer of MinervaX, said: “We are very pleased with the progress in clinical development of our maternal GBS vaccine since closing our last financing round in December 2020. The vaccine has to date demonstrated high immune responses even in individuals with low levels of preexisting immunity to GBS who are most at risk of invasive disease. The vaccine has also demonstrated a very promising safety profile in both non-pregnant and pregnant women in past and ongoing clinical studies. The progress represents a significant advancement towards initiating Phase 3 clinical studies”
“We are also very pleased to welcome Gerd Zettlmeissl as new Chairman of the Board of Directors as well as the members of the expanded Scientific Advisory Board. The additions bring valuable business and development expertise to the Company.”
Gerd Zettlmeissl, Chairman of MinervaX Board of Directors said: “I am very excited about the outstanding progress MinervaX is making in the development of one of the globally most promising GBS vaccine candidates and about the opportunity to support the company towards future success.”
Ralf Clemens, MinervaX Scientific Advisory Board member said: “Group B Streptococcus is a leading cause of neonatal sepsis and despite efforts since many years by academia and industry to develop a prophylactic vaccine progress so far is limited. The MinervaX phase 2 candidate has shown very promising immunogenicity data and an excellent safety profile, and the company has a clear plan of path to licensure. The studies in Uganda, UK and Denmark are a critical piece of that plan.”
ENDS
Enquiries
For more information on MinervaX, please contact:
Per Fischer, CEO: pbf@minervax.com

Funding to advance the company’s SMiRNA™ platform to identify clinical candidates in myotonic dystrophy type 1 (DM1), amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD) and tauopathies.
September 29, 2021 07:00 AM Eastern Daylight Time
BOSTON--(BUSINESS WIRE)--Expansion Therapeutics, Inc., a biotechnology company focused on developing transformative oral medicines for severe RNA-mediated diseases, today announced the close of an $80 million Series B financing. Cormorant Asset Management led the financing with participation from new investors Westlake Village BioPartners, Surveyor Capital (a Citadel company) and Logos Capital as well as Series A investors RA Capital Management, 5AM Ventures, Kleiner Perkins, Sanofi Ventures and Novartis Venture Fund. Proceeds from the financing will be used to advance the company’s small molecule RNA platform (SMiRNA™) to identify clinical candidates in myotonic dystrophy type 1 (DM1), amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD) and various tauopathies.
“Drugging RNA with small molecules has the potential to dramatically transform the lives of patients and we are excited to be supported by world-class investors who share our commitment to patients and our vision to develop innovative therapies to treat severe RNA-mediated diseases, including neurodegenerative and neurological disorders,” said Renato Skerlj, Ph.D., President and Chief Executive Officer of Expansion Therapeutics. “This financing milestone demonstrates significant investor confidence in the leadership team, the science and our mission to accelerate the preclinical and clinical development of our novel medicines utilizing our proprietary approach.”
Expansion focuses on structured RNA targets that are evolutionarily conserved or genetically demonstrated to cause disease. Disease-driving RNA structures amenable to ligand binding are identified by combining patient genetics with deep informatic analysis and then screened against Expansion’s proprietary RNA-focused libraries for small molecules that modulate the RNA target in a functionally relevant manner. Expansion’s novel structural biology SMiRNA™ platform is then utilized for the design and advancement of lead-like chemical matter that ultimately delivers an oral medicine to treat disease. Expansion is focused on neurological diseases where few treatment options are available for patients such as DM1 which is a system wide disorder affecting muscle, cardiac and brain function. A small molecule medicine that treats all aspects of the disease offers patients a significant benefit.
“Recent advances in understanding RNA structure and the science of targeting RNA with small molecules offer tremendous opportunities to transform the way RNA-mediated diseases are treated. We believe Expansion’s established focus on structured RNA targets position them as leaders in this emerging space,” said Andy Phillips, Ph.D., Managing
Director at Cormorant. “We’re extremely enthusiastic about the promise that the SMiRNA™ platform holds to deliverdisease-modifying therapies for a wide range of devastating neurological disorders, particularly neurodegenerativediseases,” added Raymond J. Kelleher, M.D., Ph.D., also Managing Director at Cormorant.
“The progression of Expansion’s lead programs, broad therapeutic potential of the platform, and the leadership team’sdemonstrated track record in the field of neuroscience compelled us to partner with this exceptionally strong group of drugdevelopers, as well as Cormorant and the rest of the investor syndicate,” said Andrew Levin, M.D., Ph.D., ManagingDirector at RA Capital Management. “We are confident in Expansion’s ability to leverage its differentiated platform topositively impact the lives of patients suffering from severe and debilitating neurological diseases who currently havelimited or no treatment options,” added Laura Tadvalkar Ph.D., Principal at RA Capital Management.
As part of its initiative to broaden its portfolio of small molecule medicines, Expansion also recently in-licensed two newresearch programs from Scripps Research, including a program targeting tau, an important driver of dementia disorders.The tau program builds on the observation that defective pre-mRNA encoding the production of tau protein is involved inthe formation of neurofibrillary tangles and has been linked to tauopathies such as Alzheimer’s disease, frontotemporaldementias, progressive supranuclear palsy and other neurodegenerative diseases.
Most recently, Expansion’s scientific founder, Matthew D. Disney, Ph.D. and colleague, Alicia Angelbello, Ph.D., wererecipients of the American Chemical Society’s Nobel Laureate Signature award for their pioneering work developing RNA-targeting medications for genetic diseases, including DM1.
About Expansion Therapeutics
Expansion Therapeutics is a biotechnology company focused on developing transformative oral medicines for severeRNA-mediated diseases including neurodegenerative disorders. Based on exclusive worldwide rights to groundbreakingresearch from the laboratory of Matthew D. Disney, Ph.D., at Scripps Research, Expansion has assembled the intellectualproperty, know-how, and proprietary enabling technologies and tools necessary to facilitate the creation of potent andspecific small molecule RNA modulators. Through this unique platform, Expansion is building a portfolio of novel RNA-targeted drug candidates with activity across a broad number of severe RNA-mediated disease indications. Headquarteredin Boston, Massachusetts, Expansion’s research facility is located in Jupiter, Florida. For more information, visitwww.expansionrx.com.
Contacts
Media:
Mike Beyer
Sam Brown Inc.
312-961-2502
mikebeyer@sambrown.com
CAMBRIDGE, Mass. – September 14, 2021 – ROME Therapeutics, a biotechnology company harnessing the power of the repeatome for drug development, today announced the completion of a $77 million Series B financing led by new investor Section 32. In addition, new investors Sanofi Ventures, Casdin Capital, Andreessen Horowitz and Alexandria Venture Investments participated in the round, alongside existing investors ARCH Ventures, GV and Mass General Brigham Ventures (formerly Partners Innovation Fund). Concurrent with the financing, Steven J. Kafka, Ph.D., managing partner at Section 32, and Jim Trenkle, Ph.D., head of investments at Sanofi Ventures, were appointed to ROME’s Board of Directors.
ROME’s mission is to harness the power of the repeatome – the roughly 60% of the human genome consisting of repetitive sequences of nucleic acids, known as repeats – to discover powerful new classes of medicines for cancer and autoimmune diseases. Repeats have long been considered part of the “dark genome,” but recent discoveries have demonstrated that the repeatome has pathological consequences in cancer, autoimmune disease and other therapeutic areas. Since launching in April 2020, ROME has identified multiple repeatome-derived targets and created an internal data science platform, called repeatomics, to analyze vast quantities of data available through the repeatome.
“In the short time since our founding, ROME has made significant progress uncovering the role and mechanism of the repeatome in cancer and autoimmune disease, which has led to our foundational repeatomics platform and identification of multiple tractable targets for drug discovery,” said Rosana Kapeller, M.D., Ph.D., president, chief executive officer and co-founder of ROME. “With the support of this group of premier investors, we are well-capitalized and resourced to continue advancing our lead programs into the clinic, expanding our pipeline of repeatome-derived programs and enhancing our repeatomics platform, as we seek to revolutionize the way cancer and autoimmune diseases are treated.”
“Traditional genomics has transformed how we think about drug development, diagnosis and treatment; however, for too long we have been unable to extend this approach to the ‘dark genome,’” said Dr. Kafka. “Leveraging the deep insights garnered by ROME’s experienced team of drug hunters and data scientists, ROME has developed innovative tools and techniques to generate novel insights from the repeatome in order to bring forward an entirely new and important class of disease-modifying medicines.”
About New Directors
Steven J. Kafka, Ph.D., is a managing partner at Section 32, a venture capital fund investing at the frontiers of technology and healthcare. Dr. Kafka serves as chairman of the board of Glympse Bio and a director at ImmuneID, as well as CEO and director of DA 32 Life Science Tech Acquisition Corp. Previously, Dr. Kafka served as both founding CEO and executive chairman of Thrive Earlier Detection, which was acquired by Exact Sciences in January 2021. Dr. Kafka also served as executive chairman of ArcherDX, Inc., which was acquired by Invitae in October 2020. Earlier, Dr. Kafka served as president and chief operating officer of Foundation Medicine, Inc., which was acquired by Roche in 2018, and held executive roles at multiple public and private oncology drug discovery and development companies. Dr. Kafka earned a Ph.D. in political economy and government from Harvard University and a B.A. in economics and political science from Stanford University.
Jim Trenkle, Ph.D., is currently head of investments at Sanofi Ventures and has a background in research and development, commercialization and early-stage biotech investing. Dr. Trenkle currently serves on the board of directors at Therini Bio, Glycomine and Veralox Therapeutics. Prior to joining Sanofi, Dr. Trenkle was part of the early team at Pivotal bioVenture Partners in San Francisco, where he served as Board observer for Entasis (ETTX), Inozyme (INZY), Akouos (AKUS), Fusion Pharmaceuticals (FSUN) and Karuna Therapeutics (KRTX). He began his career with Gilead Sciences where he held positions of increasing responsibility in medicinal chemistry, project and portfolio management and commercial strategy, largely focused on hepatitis C and other liver diseases. Dr. Trenkle holds a B.S. in honors chemistry from the University of Michigan, a Ph.D. in organic chemistry from Massachusetts Institute of Technology and an MBA from the University of California Berkeley, Haas School of Business.
About ROME
ROME Therapeutics is developing novel therapies for cancer and autoimmune diseases by harnessing the power of the repeatome – vast stretches of uncharted genetic material that have long been dismissed as “junk DNA.” With several drug targets identified and multiple discovery programs underway, ROME is moving rapidly to leverage this new frontier in biology. To lead this exploration, ROME has assembled a team of world-class leaders across fields including oncology, immunology, virology and machine learning. ROME is based in Cambridge, Mass. For more information, please visit www.rometx.com.
Media Contact
Lisa Qu
Ten Bridge Communications
678-662-9166
lqu@tenbridgecommunications.com
Investor Contact
Monique Allaire
THRUST Strategic Communications
monique@thrustsc.com
###
Industry leader with expertise in building successful biotechnology companies
Copenhagen, Denmark and Boston, US 13th September 2021 – Muna Therapeutics (“Muna”), pioneering the development of first-in-class small molecule therapeutics for neurodegenerative diseases, today announced the appointment of Dr. Donald Nicholson, as independent Chair of its Board of Directors.
Dr. Nicholson has deep experience across both biotech and large pharma. He was previously the CEO of Nimbus Therapeutics and was responsible for major transactions with Gilead, Celgene and Genentech. Prior to this, he spent 25 years at Merck, where he held various strategic, leadership and operational roles across diverse therapeutic areas including inflammation, immunology and neuroscience, amongst others. Dr. Nicholson began his career at Merck Frosst in Montreal as a senior research biologist and advanced through various positions of increasing responsibility, including vice president and site head of the Merck Neurosciences Research site in San Diego, vice president of immunology and infectious diseases and vice president and worldwide discovery head for the Bone, Respiratory, Immunology and Endocrine franchise based in New Jersey. He is Chair of the Board of Directors at NodThera, Disc Medicine and Jnana Therapeutics, and Board Director at Kymera Therapeutics (KYMR) and Generation Bio (GBIO).
Rita Balice-Gordon, Chief Executive Officer of Muna Therapeutics, said: “We are delighted to welcome Dr. Nicholson as independent Chair at Muna. He brings to Muna a wealth of experience in biopharma and biotech, insights into building high-functioning companies and boards, and a commitment to advancing therapies for patients with neurodegenerative diseases. We look forward to working with him and the Board as we move forward.”
Morten Graugaard Døssing, prior Chair of the Board of Directors of Muna Therapeutics said: “We are very pleased to welcome Dr. Nicholson to our Board of Directors as independent Chair. He brings to Muna his exceptional experience and leadership to support our mission to advance and strengthen our therapeutic pipeline.”
Donald Nicholson, incoming independent Chair of the Board of Directors of Muna Therapeutics said: “I am delighted to be joining Muna as Chair of the Board. Muna’s innovative approach to neurodegenerative disease therapeutics as well as its stellar leadership and scientific teams inspire great confidence in its future success.”
Dr. Nicholson has co-authored more than 150 publications in peer-reviewed scientific and medical journals and is internationally recognized for his contributions to the field of apoptotic cell death. He received his doctorate in biochemistry from the University of Western Ontario and trained as a Medical Research Council postdoctoral fellow at the University of Munich in Germany. He is the recipient of multiple academic and professional honors.
Muna Therapeutics previously raised US$ 73 million (EUR 60 million) in a Series A financing round co- led by Novo Holdings, Sofinnova Partners, Droia Ventures and LSP Dementia Fund, with Polaris Partners, Polaris Innovation Fund, Sanofi Ventures, V-Bio Ventures and VIB joining the round. The
recent raise will help Muna progress its innovative small molecule candidates to the clinic, further
develop its innovative drug discovery platform as well as expand its US presence.
ENDS
For more information please contact:
Muna Therapeutics
Rita Balice-Gordon, CEO
Email: balicegordon@munatherapeutics.com
Optimum Strategic Communications
Mary Clark, Stella Lempidaki, Zoe Bolt
Tel: +44 (0) 20 3922 1906
Email: muna@optimumcomms.com
About Muna Therapeutics
Muna Therapeutics is a private biopharmaceutical company founded in 2020 and based in Copenhagen, Denmark and Leuven, Belgium. Muna discovers and develops therapies that slow or stop devastating neurodegenerative diseases including Alzheimer’s, Frontotemporal Dementia and Parkinson’s. These disorders impact memory, movement, language, behavior and personality resulting in disability and death of millions of patients around the globe. We focus our groundbreaking science on identifying new medicines to preserve cognition and other brain functions and enhance resilience to neurodegenerative diseases. Our name reflects this focus: Muna means ‘to remember’ in Old Norse. www.munatherapeutics.com
- Financing co-led by Novo Holdings, Sofinnova Partners, Droia Ventures and LSP Dementia Fund, with participation from Polaris Partners, Polaris Innovation Fund, Sanofi Ventures, V-Bio Ventures and VIB
- Developing small molecules to repair neuronal dysfunction and resolve neuroinflammation
- Innovative drug discovery platform leveraging insights from novel targets and pathways, resilience to neurodegeneration and all-in- human validation
Copenhagen, Denmark 30 June 2021 – Muna Therapeutics (“Muna”), pioneering the development of first-in-class small molecule therapeutics for neurodegenerative diseases, today announced the successful closing of a US$ 73 million (EUR 60 million) Series A financing round. The investor syndicate was co-led by Novo Holdings, Sofinnova Partners, LSP Dementia Fund, and Droia Ventures, with Polaris Partners, Polaris Innovation Fund, Sanofi Ventures, V-Bio Ventures and VIB joining the round.
Muna is the combination of two innovative European start-up companies. Muna was founded in 2020 by progranulin pathway thought leaders Professor Simon Glerup and his team from Aarhus University, Denmark, with investor Novo Holdings. Muna developed a strategic partnership on additional targets with Axxam SpA, based in Milan, Italy. Muna is part of Novo Seeds’ company creation efforts, where the Novo team and its entrepreneurs-in-residence help build new biotech companies based on groundbreaking new science.
Muna joined forces with K5 Therapeutics, co-founded in 2020 by Professor Bart De Strooper from VIBKU Leuven, Belgium, a pioneer in neurodegenerative diseases research, with investors Droia Ventures and VIB. The combined company - Muna Therapeutics - will be based in Copenhagen and Leuven and is led by seasoned pharma executives CEO Rita Balice-Gordon and COO Anders Hinsby, both
entrepreneurs-in-residence of Novo Seeds.
Neurodegenerative diseases affect millions of individuals, with increasing global impact as the population ages. Palliative treatments are scarce, and no curative therapies are currently available. Muna is focused on addressing the staggering unmet need experienced by patients around the world with neurodegenerative disorders.
Muna’s innovative all-in-human target discovery and validation platform is based on proprietary insights into molecular pathways in different human brain cell types that underlie disease pathology and resilience to neurodegeneration, based on work from the De Strooper and Glerup laboratories. Muna has built a cutting-edge small molecule drug discovery engine that leverages high-resolution target structural approaches, AI-driven computational chemistry and cell-based screening. The financing will be used to advance Muna’s small molecule programs focused on repairing neuronal dysfunction, resolving neuroinflammation and restoring neuroprotection and resilience to disease to Investigational New Drug (IND) applications.
“We are in an era of rapid advancement in understanding how to slow or stop the relentless progression of neurodegenerative diseases like Alzheimer’s and Frontotemporal Dementia that devastate cognition and quality of life of patients as well as caregivers.” said Rita Balice-Gordon, Chief Executive Officer of Muna Therapeutics. “Our team is committed to leveraging our collective expertise to deliver impactful disease modifying small molecule therapeutics to patients as rapidly as possible.”
The Board of Directors includes: Morten Graugaard Døssing, Partner at Novo Holdings, current Chairman; Henrijette Richter, Managing Partner at Sofinnova Partners; Cillian King, Investment Manager at LSP; Luc Dochez, Partner at Droia Ventures; Isaac Ciechanover, Partner at Polaris Partners; Laia Crespo, Head of Investments at Sanofi Ventures; and Rita Balice-Gordon, CEO of Muna.
Morten Graugaard Døssing, Chairman of the Board and Partner at Novo Holdings, said: “Novo Seeds is delighted to welcome a global syndicate of first-class investors who strongly believe in Muna’s world-leading science, experienced leadership team, and its potential to develop innovative treatments for neurodegenerative diseases. We are honored to co-lead this round with Sofinnova Partners, Droia Ventures and LSP Dementia Fund – a tremendous joint effort to bring Muna to the next level.”
For more information please contact:
Muna Therapeutics
Rita Balice-Gordon, CEO
Email: balicegordon@munatherapeutics.com
Optimum Strategic Communications
Mary Clark, Manel Mateus, Stella Lempidaki
Tel: +44 (0) 20 3922 1906
Email: muna@optimumcomms.com
About Muna Therapeutics
Muna Therapeutics is a private biopharmaceutical company founded in 2020 and based in Copenhagen, Denmark. Muna discovers and develops therapies that slow or stop devastating neurodegenerative diseases including Alzheimer’s, Frontotemporal Dementia and Parkinson’s. These disorders impact memory, movement, language, behavior and personality resulting in disability and death of millions of patients around the globe. We focus our groundbreaking science on identifying new medicines to preserve cognition and other brain functions and enhance resilience to neurodegenerative diseases. Our name reflects this focus: Muna means ‘to remember’ in Old Norse. www.munatherapeutics.com
Investor Syndicate
About Novo Holdings
Novo Holdings A/S is a private limited liability company wholly owned by the Novo Nordisk Foundation. It is the holding company of the Novo Group, comprising Novo Nordisk A/S and Novozymes A/S, and is responsible for managing the Novo Nordisk Foundation’s assets. Novo Holdings is recognized as a world-leading life science investor with a focus on creating long-term value. As a life sciences investor, Novo Holdings provides seed and venture capital to developmentstage companies and takes significant ownership positions in growth and well-established companies. Novo Holdings also manages a broad portfolio of diversified financial assets. https://www.novoholdings.dk/
About Sofinnova Partners
Sofinnova Partners is a leading European venture capital firm in life sciences, specializing in healthcare and sustainability. Based in Paris, London and Milan, the firm brings together a team of professionals from all over the world with strong scientific, medical and business expertise. Sofinnova Partners is a hands-on company builder across the entire value chain of life sciences investments, from seed to later-stage. The firm actively partners with ambitious entrepreneurs as a lead or cornerstone investor to develop transformative innovations that have the potential to positively impact our collective future. Founded in 1972, Sofinnova Partners is a deeply-established venture capital firm in Europe, with 50 years of experience backing over 500 companies and creating market leaders around the globe. Today, Sofinnova Partners has over €2 billion under management. www.sofinnovapartners.com
About Droia Ventures
Droia is a specialist biotech investor with an exclusive focus on therapeutics for oncology and genetic disease. Droia invests globally in newly founded or early-stage platform companies that apply novel science and innovative technologies to bring first-in-class drug candidates to patients. With our team of seasoned scientists, entrepreneurs and investment professionals we build great companies to save patient lives. www.droiaventures.com.
About LSP Dementia Fund
LSP is one of the largest European investment firms providing financing for life sciences and health care companies. LSP’s management has raised over €2 billion ($2.5 billion) and supported the growth of 300 companies since it started to invest in 1988, including signature deals such as argenx, Crucell and Neuravi. With offices in Amsterdam, Munich and Boston, LSP currently has the possibility to invest through five strategies, each having a distinctive investment scope and a dedicated team: LSP 6 invests in private early- to late-stage drug development and medical technology companies; LSP HEF 2 focuses on private late-stage medical technology companies; the LSP Dementia Fund invests in companies targeting neurodegenerative diseases; LSP Public targets public healthcare companies; and EBAC is the first healthcare SPAC to exclusively focus on European biotech. LSP is an active contributor to the global life sciences industry and the European life science eco-system by assuming for-profit and notfor- profit roles as initiators, founders and board members in various private and public bodies and organizations, for example being founder and board member of the Oncode Institute. www.lspvc.com.
About Polaris Partners and Polaris Innovation Fund
Polaris Partners has a 20-plus-year history of partnering with repeat entrepreneurs and world-class innovators who are improving the way we live and work. The multibillion-dollar firm manages specialty and diversified funds in healthcare and technology with investments across all stages. The Polaris Innovation Fund aims to accelerate the commercial and therapeutic potential of early-stage academic research. By partnering with passionate entrepreneurs with transformational science, the Polaris Innovation Fund fosters company creation and growth through an active investment model. Polaris has offices in Boston, San Francisco, and New York. www.polarispartners.com.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi. Sanofi Ventures invests in early-stage biotech and digital health companies with innovative ideas and transformative new products and technologies of strategic interest to Sanofi. Among these areas are oncology, immunology, rare diseases, vaccines, potential cures in other core areas of Sanofi’s business footprint, and digital health solutions. www.sanofiventures.com
About V-Bio Ventures
V-Bio Ventures is an independent venture capital firm specialized in building and financing young, innovative life science companies. V-Bio Ventures was established in 2015 and works closely with Belgium-based VIB, one of the world’s premier life science institutes. The fund invests throughout Europe in start-up and early-stage companies with high growth potential focusing on technologies that provide transformational improvements in the biopharmaceutical, pharmaceutical, diagnostics and agricultural sectors. www.v-bio.ventures
About VIB
VIB is an entrepreneurial research institute in life sciences located in Flanders, Belgium. VIB’s basic research leads to new and innovative insights into normal and pathological life processes. It unites the expertise of all its collaborators and research groups in a single institute, firmly based on its close partnership with 5 Flemish universities (Ghent University, KU Leuven, University of Antwerp, Vrije Universiteit Brussel and Hasselt University). We are supported by a solid funding program from the Flemish government. VIB has an excellent track record on translating basic scientific results into pharmaceutical, agricultural and industrial applications. Since its foundation in 1996, VIB has created 29 start-up companies, now employing over 900 people. www.vib.be
SAN CARLOS, Calif., June 29, 2021 – Glycomine, Inc., a biotechnology company focused on developing new therapies for orphan diseases, today announced the publication in Molecular Genetics and Metabolism of a key finding from a natural history study of phosphomannomutase 2-congenital disorder of glycosylation (PMM2- CDG), a rare pediatric orphan disease. In this study, the levels of morning cortisol and adrenocorticotropic hormone (ACTH) were measured in a cohort of patients and found to be significantly below normal, indicating PMM2-CDG patients are at risk for secondary adrenal insufficiency. These data provide key insights to improve standard of care, as early recognition of adrenal insufficiency and initiation of glucocorticoid replacement therapy and stress dosing could be lifesaving. The authors conclude that morning cortisol and ACTH levels should be evaluated at least annually for all patients with PMM2-CDG. If abnormal, a low dose ACTH stimulation test should follow to evaluate the hypothalamus, pituitary, adrenal (HPA) axis.
“Endocrinopathies in PMM2-CDG have not received the attention they deserve, especially considering that glycoproteins are involved in virtually every endocrine axis,” said Kyriakie Sarafoglou, M.D., Associate Professor in the Department of Pediatrics at University of Minnesota and lead author of the paper. “Through an international collaboration, this study was the first to identify that patients with PMM2-CDG are at risk for secondary adrenal insufficiency and to suggest that morning cortisol and ACTH monitoring should become part of standard care in these patients.”
The natural history study completed enrollment with 139 PMM2-CDG patients at 11 sites around the world (ClinicalTrials.gov Identifier: NCT03173300). Morning serum cortisol and ACTH levels were simultaneously measured in a cohort of 43 patients. In this cohort, 11 patients (25.6%) had cortisol below 5 μg/dl and low to normal ACTH levels, suggestive of secondary adrenal insufficiency. Secondary adrenal insufficiency has a prevalence of 1.5 to 2.8 in 10,000 (< 0.03%) in the general population. Since this finding, two of the 11 patients have been confirmed to have central adrenal insufficiency after low dose ACTH stimulation test results and are on hydrocortisone replacement and/or stress dosing during illness.
“We initiated this natural history study, which is the largest longitudinal study ever undertaken for this disease, to learn more about how PMM2-CDG affects patients and inform the design of our clinical trials for GLM101,” said Horacio Plotkin, M.D., FAAP, Chief Medical Officer of Glycomine. “Through thoughtfully and scientifically studying the clinical presentation and biochemistry of PMM2-CDG, our investigators have uncovered a significant risk factor that is often masked by other co-morbidities of the disease. We thank the investigators and the patients and families who participated in this study for their time and support as we advance into clinical trials GLM101. Good things happen when patients and families, investigators, and companies work together.”
“Even before we have started our first interventional clinical trial, we have uncovered a potential life-saving therapeutic intervention,” said Peter McWilliams, Ph.D., CEO of Glycomine. “While cortisol/ACTH monitoring and glucocorticoid therapy have the potential to be lifesaving for PMM2-CDG patients during stress or illness, they are still only addressing the symptoms of the disease. We look forward to initiating later this year our interventional clinical trials for GLM101 that has the potential to restore the deficiency in mannose-1-phosphate that causes PMM2-CDG.”
The research entitled, “Should patients with Phosphomannomutase 2-CDG (PMM2-CDG) be screened for adrenal insufficiency?” is available at this link: https://doi.org/10.1016/j.ymgme.2021.06.003.
SAN CARLOS, Calif., June 23, 2021 – Glycomine, Inc., a biotechnology company focused on developing new therapies for orphan diseases, today announced that it has closed a $68 million Series B financing. The proceeds of the financing will be used to advance Glycomine’s lead drug candidate, GLM101, through initial clinical trials in patients. GLM101 is a novel substrate replacement therapy in development to treat phosphomannomutase 2- congenital disorder of glycosylation (PMM2-CDG), a rare disease representing a critical unmet medical need.
The Series B financing includes $35 million of new funds in addition to the $33 million announced in August 2019. Today’s financing was led by new investors, Abingworth and Sanofi Ventures, and joined by RiverVest Venture Partners and Remiges Ventures. In addition, all previous Series B investors – Novo Holdings A/S, Asahi Kasei Pharma Ventures, Mission BioCapital, Sanderling Ventures, and Chiesi Ventures – participated.
Glycomine’s CEO, Peter McWilliams, Ph.D., said, “We are delighted to have expanded our syndicate with these high-quality, experienced life science investors. We have demonstrated in preclinical studies that GLM101 can restore the glycosylation pathways that are disrupted in PMM2-CDG. This additional funding will enable us to confirm in the clinic the potential of GLM101 as a therapy for all PMM2-CDG patients, regardless of genotype, and we are looking forward to executing on our clinical program with this new infusion of capital.”
PMM2-CDG is the most prevalent congenital disease of glycosylation but has no FDA-approved treatments. Glycomine’s GLM101 is a mannose-1-phosphate replacement therapy in development to treat PMM2-CDG, a disease caused by a deficiency of the enzyme phosphomannomutase 2 (PMM2). PMM2 converts mannose-6- phosphate to mannose-1-phosphate, which is an essential sugar molecule in the N-glycosylation pathway and is crucially important for proper glycoprotein structure and function. GLM101 is designed to deliver mannose-1- phosphate directly into cells and thereby bypass the PMM2 enzyme deficiency and address all disease-causing PMM2 mutations to restore pathway function. GLM101 has received Orphan Drug Designation in the U.S. and Europe and Rare Pediatric Disease Designation in the U.S.
“Glycomine’s novel and scientifically informed approach shows the potential of GLM101 to be a significant disease-modifying treatment,” added Bali Muralidhar, M.D., Ph.D., Managing Partner with Abingworth. “Glycomine has developed a proprietary approach to delivering mannose-1-phosphate intracellularly to replace the missing sugar molecule. Initial clinical studies of GLM101 will be an important proof-of-concept, and we are excited to support the company at this pivotal stage.”
“PMM2-CDG is an important unmet need with no therapeutic option,” added Jim Trenkle, Ph.D., US Head of Investments at Sanofi Ventures. “We are enthusiastic to support Glycomine’s work to advance transformative therapies for PMM2-CDG patients and their families and caregivers.”
In connection with the financing, Dr. Muralidhar, Dr. Trenkle, and Niall O’Donnell, Ph.D., Managing Director of RiverVest, have been appointed to Glycomine’s Board of Directors.
FREDERICK, Md – June 16, 2021 – Veralox Therapeutics, a biotechnology company developing first-in-class small molecule therapeutics that treat the underlying pathologies of diseases with significant unmet medical needs, today announced the closing of a $16.6 million Series A financing. The round was led by Hatteras Venture Partners with participation from Genesys Capital, Point Field Partners and Alexandria Venture Investments as well as support from previous Veralox investors including Sanofi Ventures, JDRF T1D Fund, Maryland Momentum Fund, VTC Innovation Fund and TEDCO. The Company also announced that Ben Scruggs, PhD, principal at Hatteras Venture Partners, and Jamie Stiff, MBA, managing director at Genesys Capital, will join the Veralox Therapeutics board of directors.
“In recent months, we have achieved significant momentum in our goal to build a robust pipeline of therapeutics targeting 12-lipoxygenase, which is associated with the onset and progression of a range of serious diseases and conditions,” said Jeffrey W. Strovel, PhD, chief executive officer of Veralox Therapeutics. “We are very pleased to have leading life sciences investors participating in our Series A financing, which will support our efforts to advance the development program for our lead product candidate VLX-1005 for the treatment of heparininduced thrombocytopenia (HIT).”
In January 2021, Veralox announced that the U.S. Food and Drug Administration (FDA) granted Orphan Drug Designation (ODD) for VLX-1005, a first-in-class small molecule inhibitor of 12- lipoxygenase, for the treatment of HIT. The FDA also approved the company’s Investigational New Drug (IND) application for initiation of a Phase 1 clinical trial of VLX-1005 in HIT.
“Veralox’s approach has the potential to be a major advance for the treatment of heparininduced thrombocytopenia, and we believe targeting the underlying pathology holds promise to improve clinical outcomes for these patients,” said Ben Scruggs, PhD, principal at Hatteras Venture Partners. “Hatteras is delighted to support Veralox’s efforts to develop novel therapeutics for this area of significant clinical unmet need.”

Milpitas, CA, June 15 Milpitas, CA, June 15, 2021 – Bigfoot Biomedical announced today that it has acquired the intellectual property assets of Common Sensing, a Cambridge, MA-based company that develops and manufactures data-driven hardware and software solutions for people using injectable medicine, including Gocap.
“This acquisition will accelerate the innovative development work already underway at Bigfoot as we advance and expand our insulin-delivery portfolio,” said Jeffrey Brewer, CEO of Bigfoot Biomedical. “There’s tremendous momentum for smart and connected technology within the diabetes industry, and Bigfoot Biomedical is uniquely poised to deliver solutions that are simple, convenient and easy to use.”
“We’ve admired the maverick spirit of the Bigfoot Biomedical team for many years as both our companies have worked incredibly hard to develop solutions for the more than 20 million people using injectable medicine in the US and EU” said Kevin Schmid, CEO of Common Sensing. “Bigfoot has 6/16/21, 3:31 PM Page 2 of 9 proven itself as a company that can gain regulatory approval and effectively lead a manufacturing and implementation strategy. This is important as we fully expect the number of people needing to intensify their insulin therapy with multiple daily injections to grow especially with the advent of simple and accessible technologies.”
Last month, Bigfoot received FDA clearance for its Bigfoot Unity™ Diabetes Management System featuring first-of-its-kind smart pen caps for insulin pens used to treat Type 1 and Type 2 diabetes. Integrated with Abbott’s FreeStyle® Libre 2 system, the Bigfoot Unity System is the first and only solution to translate continuously monitored glucose data into on-demand insulin dose recommendations, based on HCP instructions, displayed right on the pen-cap screen for ease of use.
“For the millions of people with insulin-requiring Type 1 and Type 2 diabetes, there’s a huge need for solutions that are accessible in terms of cost and simplicity,” said Brewer. “We’re keeping our foot firmly on the gas pedal as we look to expedite the 6/16/21, 3:31 PM Page 3 of 9 incredible work being done currently by the Bigfoot team. Making smart acquisitions that are compatible with our mission and philosophy are key to our strategy of delivering valued innovations to the diabetes community.”
BOSTON and NEW YORK, May 11, 2021 /PRNewswire/ -- Health care technology company Aetion announced today a $110 million Series C fundraise, led by Warburg Pincus, a leading global growth equity rm, with additional investments from B Capital and Foresite Capital. Aetion's existing backers New Enterprise Associates (NEA) and Flare Capital Partners also joined the round. Aetion has raised a total of $212 million to date. The capital infusion supports Aetion's continued growth and industry leadership in the real-world evidence (RWE) space. In the last year alone, Aetion nearly doubled revenue and secured 100 percent of customer renewals while establishing valuable industry collaborations. Aetion became the rst RWE company to establish an FDA COVID-19 research collaboration agreement and was selected by ICER as its preferred RWE partner and platform.
"In a post-COVID era, we have an imperative to use rapid, rigorous evidence to inform health care decisions," said Aetion CEO, Carolyn Magill. "This investment reinforces Aetion's position as an RWE leader and our potential for future growth as we enable our customers to generate regulatory-grade evidence at scale."
The company will use the new funds to extend the capabilities of its Aetion Evidence Platform®, expand its European and Asian-Pacic footprint, and grow its commercial team in order to serve the growing demand from biopharma, payer, and medical device / diagnostics customers.
Just a few years after its launch, the Aetion Evidence Platform is used by the majority of the top global biopharma rms, leading payers, and regulatory and HTA agencies to inform decisions on the safety, effectiveness, and value of medical products.
Warburg Pincus is a leading investor in health care and technology, with signicant expertise in venture capital and growth investing. Since its inception, the rm has invested in excess of $12 billion and $22 billion into health care and technology companies, respectively. Current health care and health care IT investments include, Alignment Healthcare, Experity, Intelligent Medical Objects, Modernizing Medicine, Outset Medical, Qualifacts, Quantum Health, SOC Telemed, Summit Health, and WebPT.
"Aetion has shown incredible growth over the last year, scaling its platform and forming industry-leading partnerships. As the use of real-world evidence expands globally, we believe our investment will help advance Aetion's technology and further inect its growth," said T.J. Carella, Managing Director and Head of Healthcare at Warburg Pincus. "We are excited to back this excellent management team and platform to further accelerate the use of data in healthcare," added Amr Kronfol, Managing Director at Warburg Pincus.
Aetion's growth has been accompanied by recognition of its status as a premier talent destination. Last year, the company was named to Fast Company's top 10 Best Workplaces for Innovators and BuiltIn's Best Places to Work in New York City and Boston; Aetion CEO Carolyn Magill was named to Rock Health's 2021 Top 50 in Digital Health; and Co-Founder, President, and Chief Science Ofcer, Jeremy Rassen, was cited among FiercePharma's Most Inuential People in the Fight Against COVID-19. Also in 2020, Cathy Polinsky, technology leader at Shopify and former StichFix CTO, joined Aetion's board of directors.
SOUTH SAN FRANCISCO, Calif, May 10, 2021 /PRNewswire/ -- Therini Bio, Inc. announced it has completed an oversubscribed Seed Extension round of nancing. The capital will accelerate the development of Therini's lead program – a monoclonal antibody against brin – towards the clinic for patients with inammatory conditions associated with vascular damage.
Therini Bio was co-founded by Dr. Katerina Akassoglou, PhD, based upon discoveries from her laboratory at Gladstone Institutes and also UC San Francisco that a cryptic epitope on brin, a blood-clotting factor, drives toxic chronic inammation in the brain and the invention of an antibody to selectively target this brin cryptic epitope. This mechanism is believed to underlie damage in multiple neurodegenerative diseases including Alzheimer's disease and Multiple Sclerosis, in addition to peripheral inammatory conditions like colitis and kidney disease. The scientic team at Therini led by Chief Scientic Ofcer Dr. Jeff Stavenhagen, PhD, has advanced this pioneering work and developed an optimized therapeutic antibody appropriate for human use that attenuates brin-induced inammation without affecting critical clotting functions. In addition, Therini also has developed a suite of antibodies that could be utilized as imaging agents for diagnosis and clinical trial selection and has also discovered a broad set of novel human therapeutic antibody candidates that it is proling for potential use in a wide range of inammatory diseases. The Company is using the capital from this nancing for antibody manufacturing and IND enabling studies in anticipation of initiating human clinical trials in 2022.
"The upsized nancing validates the progress made by Therini's research team and reveals the signicant excitement that exists around the breadth and depth of the science behind this new biological approach to inammation. Having such prestigious corporate and institutional investors around the table provides the critical nancial and human capital needed to support Therini through this stage of development as we near human clinical trials. The round will allow us to begin developing our exciting portfolio of antibodies to treat and diagnose a wider array of clinical indications," stated Dan Burgess, President and CEO of Therini. The round was co-led by SV Health Investors' Impact Medicine Fund, MRL Ventures, and Sano Ventures who join existing investors including the Dementia Discovery Fund and Dolby Family Ventures, as well as new investor, Foundation for a Better World in support of the Company. With the closing of the nancing Dr. Christine Brennan, PhD from MRL Ventures and Dr. Jim Trenkle, PhD from Sano Ventures have joined the Therini board of directors.
LOS ANGELES & NEW YORK--(BUSINESS WIRE)--Science 37, Inc. (“Science 37”), developer of the Decentralized Clinical Trial Operating System™, and LifeSci Acquisition II Corp. (NASDAQ: LSAQ) (“LifeSci”), a blank check company targeting the biopharma, medical technology, digital health and healthcare services sectors, announced today that they have entered into a definitive business combination agreement. Upon closing of the proposed transaction, the combined company will operate as Science 37 and is expected to be listed on the NASDAQ under the ticker symbol “SNCE”. The proposed transaction values Science 37 at an initial enterprise value of approximately $1.05 billion and will provide the combined company with approximately $250 million of cash (assuming no redemptions from LifeSci’s trust account), to fuel continued growth.
“The clinical research industry is undergoing a dramatic transformation in which traditional development methods are being supplanted by technology fueled innovation,” said David Coman, Chief Executive Officer of Science 37. “Our clinical trial Operating System (OS) can enable significantly faster enrollment, retain patients at a meaningfully higher rate, and achieve higher enrollment among diverse patient populations. With this investment, we expect to advance our OS to further penetrate adjacent markets, and power the future of clinical research where we bridge between the traditional and decentralized approaches to enable a truly Agile Clinical Trial.”
Andrew McDonald, Ph.D., Chief Executive Officer of LifeSci Acquisition II Corp, said, “healthcare is increasingly transitioning to virtual and home-based environments, and we believe Science 37 is uniquely positioned as a pioneer in its approach to clinical trials. The company’s rapid growth is a testament to its truly disruptive technology and its immense market opportunity to change the way drugs are developed and go to market.”
SEATTLE--(BUSINESS WIRE)--Icosavax, Inc. today announced the close of a $100 million Series B Financing, led by RA Capital Management and joined by Janus Henderson Investors, Perceptive Advisors, Viking Global Investors, Cormorant Asset Management, Omega Funds, and Surveyor Capital (a Citadel company). Icosavax’s existing investors also participated, including Qiming Venture Partners USA, Adams Street Partners, Sanofi Ventures, and ND Capital. A previously announced funding from Open Philanthropy was included in this round. In conjunction with the financing, Peter Kolchinsky, Ph.D., founder and Managing Partner of RA Capital Management, will join the company’s Board of Directors.
The proceeds of the financing will support the development of Icosavax’s bivalent respiratory syncytial virus (RSV) and human metapneumovirus (hMPV) vaccine program through initial clinical studies, continued evaluation of its SARS-CoV-2 vaccine candidate, and further expansion of the company’s pipeline of VLP vaccine candidates focused on protecting older adults from life-threatening respiratory diseases.
“There are currently no approved vaccines against RSV and hMPV, two respiratory viruses that are disproportionately lifethreatening for the elderly. We are excited to lead this investment in Icosavax alongside a great syndicate to advance the combination RSV/hMPV candidate through development and into the clinic,” said Peter Kolchinsky, Ph.D., Managing Partner at RA Capital Management.
“We are delighted to have attracted a top-tier investor syndicate who recognize the potential of our VLP technology to create more effective and durable vaccines for at-risk populations, like the elderly, where traditional vaccines have reduced efficacy,” said Adam Simpson, Chief Executive Officer of Icosavax. “Based on preclinical data, we believe our vaccine candidates could offer significant protection against leading viral causes of pneumonia in older adults where no licensed vaccines currently exist.”
The company’s RSV vaccine candidate, IVX-121, incorporates a stabilized prefusion F antigen licensed from NIAID/NIH (DS-Cav1; Science 2019). In a Phase 1 clinical study conducted by NIAID/NIH, DS-Cav1 was shown to induce robust neutralizing antibody titers, higher than titers shown in previous studies with RSV postfusion F vaccine candidates. Extensive preclinical studies suggest that the VLP display of DS-Cav1 in IVX-121 induces higher and more durable Icosavax Closes $100 Million Series B Financing to Advance Bivalent RSV/hMPV Vaccine Candidate Into Clinical Trials Company’s novel computationally designed virus-like particle (VLP) technology has the potential to create safe, effective, and durable vaccines against life-threatening respiratory viruses in older adults neutralizing antibody titers compared to the DS-Cav1 antigen alone, and presentation of DS-Cav1 on the VLP confers improved stability. Icosavax plans to advance IVX-121 into clinical studies this year. Data from the first in human study of IVX-121 in young and older adults will inform and enable the transition to a bivalent RSV/hMPV clinical program.
For COVID-19, Icosavax is advancing a SARS-CoV-2 receptor binding domain VLP vaccine candidate, IVX-411, into initial clinical studies in 2021. Preclinical data on IVX-411 and precursor candidates in mice and non-human primates show induction of robust neutralizing antibody titers and protection from viral challenge (Cell 2020, Preprint 2021). Data from the IVX-411 clinical study will inform potential development paths in the rapidly evolving COVID vaccine landscape.
NEW YORK--(BUSINESS WIRE)--Click Therapeutics, Inc. (“Click”), a leader in Digital Therapeutics™ as prescription medical treatments, today announced three corporate governance additions to further position the organization for continued growth and advancing its mission to develop and commercialize software as treatments. Wall Street veteran Randall Stanicky has joined the company leadership as Chief Financial Officer; accomplished digital healthcare executive Lee Shapiro has joined the Board of Directors; and former biopharmaceutical executive Muzammil Mansuri, Ph.D. has expanded his role at Click through appointment as Board Chair.
“We are honored to have these industry experts on the Click team,” said David Benshoof Klein, co-founder and CEO of Click Therapeutics. “As we discover, develop and commercialize a broad pipeline of prescription digital therapeutics for the treatment of multiple medical conditions, the experience and respected tenures of Randall, Lee, and Muz solidify Click’s leadership role in digital therapeutics and in the broader digital health space.”
Joining as CFO with more than 20 years of extensive industry experience and most recently leading RBC Capital Markets’ large cap, specialty and generic pharmaceuticals team, Randall Stanicky brings the focused financial leadership necessary to expand Click’s clinical and commercial reach. Prior to RBC, Stanicky spent the majority of his Wall Street career in a similar role at Goldman Sachs. He holds a Bachelor of Commerce degree from the University of British Columbia and sits on the Board of Directors at the Children's Tumor Foundation where he serves as an officer and chair of the audit committee. "The transformative potential of digital therapeutics and the breadth of Click's platform is what drew me to the company," said Stanicky. "With clear opportunities to scale the organization and improve clinical outcomes in millions of users, the executive team and I are primed to succeed with our vision."
Lee Shapiro joins Click’s Board of Directors and will serve as chair of the audit committee. He is Managing Partner at 7wireVentures, an investment firm he co-founded over a decade ago. He recently served as Chief Financial Officer of Livongo Health until their successful $18.5B merger with Teladoc. Prior to Livongo, Mr. Shapiro was President of Allscripts from 2001 until the end of 2012. He has also served on the Board of Directors of leading health technology companies such as Medidata, ConsejoSano, Medisafe, HomeThrive and other 7wire portfolio companies. He is a member of the National Board of Directors of the American Heart Association, where he chairs the audit committee and serves on the business operations committee. He holds a J.D. from The University of Chicago Law School and a B.S. in accounting from the University of Illinois.
Dr. Muzammil Mansuri has expanded his role from Senior Strategic Advisor to Click’s Board to Chair of the Board. With over three decades of comprehensive experience in senior roles within the global biopharmaceutical industry, Dr. Mansuri’s deep knowledge of therapeutic and digital approaches to patient care is key as Click advances an industry-leading digital therapeutics pipeline. Dr. Mansuri currently serves as Partner at F-Prime Capital. Prior to joining F-Prime, Dr. Mansuri was a member of the Executive Committee at Sanofi, as Executive Vice President, Strategy, Business Development, and Click Therapeutics Expands its Leadership with Executive and Board Appointments Licensing. Before this, he was at Gilead Sciences as Senior Vice President, Research and Development Strategy and Business Development. In that capacity he led the R&D Strategy, Business Development, and M&A activities for the company. Dr. Mansuri has served as Chief Executive Officer of several biotech companies and as a General Partner at Flagship Ventures (now Flagship Pioneering). He began his career in the pharmaceutical industry as a bench medicinal chemist with Bristol Myers. He received his B.Sc. and Ph.D. in chemistry from the University College London and conducted postdoctoral research at UCLA and Columbia University.
The executive leadership and Board announcements follow Click’s strategic partnerships with Otsuka, Boehringer Ingelheim, and a variety of payers, employers, and thought leaders.
Shapiro stated, “I have had the privilege to work with and evaluate numerous organizations seeking to advance the future of digital health. Click is uniquely positioned to seize on this moment to provide a new type of digital care. I am thrilled to join the Board to help ensure the company succeeds in this opportunity, and to become an investor in the company along with my business partner Glen Tullman.”
Board Chair Mansuri added, “Clinically validated digital therapeutics are the real solutions needed to improve patients' health outcomes beyond what we have been able to achieve with pharmacotherapy alone. In a time when obtaining access to comprehensive care can be more difficult than ever, Click is positioned to change this paradigm.”
SAN MATEO, Calif.--(BUSINESS WIRE)--Evidation Health today announced the close of $153 million in Series E growth funding to rapidly expand its virtual health programs on Achievement , the largest digital health network in the United States. The round was co-led by OMERS Growth Equity and Kaiser Permanente Group Trust; existing investors, including McKesson Ventures and B Capital Group also participated. Teresa Lee, Managing Director, OMERS Growth Equity, joins Evidation’s Board of Directors.
Evidation will use this additional capital to fuel the expansion of virtual health programs on the Achievement platform, building on its trusted relationships with individuals and its experience synthesizing and analyzing person-generated health data for stakeholders across healthcare. Evidation’s new programs will provide personalized insights and tools to motivate and empower individuals to take evidence-supported actions to manage their health and conditions.
“Achievement has made it possible to rapidly understand health and therapeutic impact with speed and rigor at scale,” said Deborah Kilpatrick, PhD, Co-CEO and Executive Chair at Evidation. “The next era of Evidation will transform how individuals interact with the ecosystem of care, by providing anyone with evidence-supported guidance and actionable tools to better understand and improve their health. We are now the first company that can help digital health and biopharma companies generate evidence about their treatments, and then enable individuals to use that evidence to take actions to manage their health.”
Comprised of more than 4 million individuals, Achievement is uniquely positioned to power virtual health at scale. The privacy-forward app and platform powering Evidation’s research business, Achievement is the largest and most geographically and demographically diverse connected cohort in the United States, representing 50 states and nine out of every 10 ZIP codes nationwide. Launched as a research platform powered by individuals and their permissioned health data, Achievement has been the basis for pioneering real-world studies across diverse topics, from COVID-19 and Alzheimer’s disease to chronic pain and respiratory conditions. Among its biopharma customers, Evidation works with nine out of the 10 largest in the world, along with academic institutions, public health organizations, and medical specialty societies. To date, Evidation has conducted more than 100 real-world studies across therapeutic areas. In 2020, the company reached the milestone of more than 1 million individuals participating in research and health programs and expects to surpass 2 million later this year.
“We are excited to support Evidation as it continues to scale its individual-centric Achievement platform,” said Teresa Lee of OMERS Growth Equity. “Evidation has made rapid, virtual research at scale possible and the company’s innovative platform can be leveraged to guide patients on their health journeys. Evidation is positioned to be a key enabler of the feedback loop that translates into timely, appropriate care and health insights.”
Harnessing its experience in research and connection to individuals through person-generated health data, last year Evidation launched its first virtual health initiatives, including Achievement for Heart Health—a first-of-its-kind health program in partnership with the American College of Cardiology to help individuals monitor and improve their cardiovascular health outside clinic-based settings, with an initial focus on heart failure. The company also collaborated with Apple and the government of Singapore on LumiHealth, a personalized program that encourages healthy activity and behaviors using Apple Watch. Using a double opt-in, these privacy-safe health programs seamlessly connect research and care, accelerating the discovery and dissemination of evidence-supported practices that improve health outcomes.
Individual privacy and per-use consents to share data will remain core tenets as the company transforms the way everyday health is understood and expands the programs available through Achievement. To join Achievement and learn more about how you can contribute to innovative health research and participate in personalized health programs, visit myachievement.com.
UTRECHT, The Netherlands and PHILADELPHIA, March 24, 2021 (GLOBE NEWSWIRE) -- LAVA Therapeutics B.V. (Nasdaq: LVTX), a biotechnology company focused on applying its expertise in bispecific gamma-delta T cell engagers to transform cancer therapy, today announced the pricing of its initial public offering of 6,700,000 common shares at a public offering price of $15.00 per share. In addition, LAVA has granted the underwriters a 30-day option to purchase up to an additional 1,005,000 common shares at the initial public offering price, less underwriting discounts and commissions. All of the shares are being offered by LAVA. The shares are expected to begin trading on the Nasdaq Global Select Market on March 25, 2021 under the ticker symbol “LVTX.”
Gross proceeds of the offering, before deducting underwriting discounts and commissions and other offering expenses payable by LAVA, are expected to be $100.5 million. The offering is expected to close on March 29, 2021, subject to the satisfaction of customary closing conditions.
J.P. Morgan, Jefferies and SVB Leerink are acting as joint book-running managers for the offering. Kempen & Co is acting as lead manager for the offering.
The registration statement relating to these securities became effective on March 24, 2021. The offering will be made only by means of a prospectus, copies of which may be obtained from J.P. Morgan Securities LLC, c/o Broadridge Financial Solutions, 1155 Long Island Avenue, Edgewood, NY 11717, by telephone at (866) 803-9204, or by email at prospectus_eqfi@jpmchase.com; Jefferies LLC, Attention: Equity Syndicate Prospectus Department, 520 Madison Avenue, 2nd Floor, New York, NY 10022, or by telephone at (877) 821-7388 or by email at prospectus_department@Jefferies.com; or SVB Leerink LLC, Attention: Syndicate Department, One Federal Street, 37th Floor, Boston, MA 02110, by telephone at (800) 808-7525, ext. 6105, or by email at syndicate@svbleerink.com.
This press release shall not constitute an offer to sell or the solicitation of an offer to buy, nor shall there be any sale of these securities in any state or jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction.
Forward-Looking Statements
This press release includes certain disclosures that contain “forward-looking statements,” including, without limitation, statements regarding LAVA’s expectations regarding the commencement of trading of its shares on the Nasdaq Global Select Market and the completion and timing of the closing of offering. Forward-looking statements are based on LAVA’s current expectations and are subject to inherent uncertainties, risks and assumptions that are difficult to predict. Factors that could cause actual results to differ include, but are not limited to, risks and uncertainties related to the satisfaction of customary closing conditions and the completion of the offering. These and other risks and uncertainties are described more fully in the section titled “Risk Factors” in the final prospectus related to the offering to be filed with the Securities and Exchange Commission. Forward-looking statements contained in this press release are made as of this date, and LAVA undertakes no duty to update such information except as required under applicable law.
Company Contact: IR@lavatherapeutics.com
Veteran biopharma CFO brings over 20 years of experience leading finance functions at publicly traded pharmaceutical companies
Utrecht, The Netherlands and Philadelphia, USA – March 15, 2021 – LAVA Therapeutics B.V. (“LAVA”, or the “Company”), a biotechnology company focused on applying its expertise in bispecific gamma-delta T cell engagers to transform cancer therapy, today announced that it is expanding its management team with the appointment of Edward F. Smith as its chief financial officer.
Mr. Smith has served on management teams of publicly traded life science companies for the past 20 years, raising approximately $500 million, building finance organizations and supporting operations from early development stage into commercialization.
“I am excited to welcome Ed to the team as LAVA prepares to become a clinical stage company,” said Stephen Hurly, chief executive officer of LAVA. “We have built a highly experienced team committed to a culture of tenacious hard work, creativity, and collaboration to drive our platform of bispecific gamma-delta T cell engagers forward to serve cancer patients. Ed fits in perfectly and brings significant experience building finance and accounting functions to our team.”
“While first generation T-cell engagers had great promise in many areas, that promise has yet to be fully realized. I believe LAVA’s approach leveraging the unique attributes of gamma delta T-cells holds the potential to move the field forward and potentially transform the standard of care across many tumor types,” Mr. Smith said.
Prior to LAVA, Mr. Smith was CFO of Marinus Pharmaceuticals and PolyMedix, Inc., and prior to that was executive director of finance at InKine Pharmaceutical Company, Inc., where he assisted with the acquisition of that company by Salix Pharmaceuticals, Inc. Earlier in his career, he held various positions of increasing responsibility in public accounting, most recently in the audit practice at Deloitte & Touche, LLP. Mr. Smith is currently a member of the board of directors at Benitec Biopharma, Inc., a development-stage biotechnology company focused on the advancement of novel genetic medicines. Mr. Smith holds a B.S. in business administration from the University of Hartford and was licensed as a Certified Public Accountant in Pennsylvania.
BOSTON, Feb. 25, 2021 /PRNewswire/ -- Medisafe, a leading digital therapeutics company creating digital drug companions, today announced that it has raised $30 million in Series C funding with investments led by Sano Ventures and ALIVE Israel HealthTech Fund, joined by Leumi Partners, Menorah Mivtachim and Consensus Business Group, previous investors Pitango Ventures, 7wireVentures, Merck Ventures, Octopus Ventures, lool HealthTech, Triventures and Ourcrowd. With this new investment Medisafe will further expand its end-to-end solutions supporting patients managing medications and accelerate revenues growth. Cris De Luca, Global Head, Digital Investments at Sano Ventures and David Klein, Co-Founder, Managing Partner ALIVE Israel HealthTech Fund will join Medisafe's board of directors.
"The COVID-19 pandemic has proven to us that supporting patients digitally through virtual care and leveraging data-driven insights, is going to be foundational to future models of healthcare and improving outcomes," said Cris De Luca, Global Head, Digital Investments at Sano Ventures.
"We've been following Medisafe and witnessed its massive growth as the healthcare industry is rapidly deploying digital solutions," said David Klein, Co-Founder, Managing Partner ALIVE Israel HealthTech Fund. "We're excited to partner with the company, its investors and partners at this inection point to drive together to new heights."
Founded in 2012, Medisafe is the largest medication management platform with over 7M registered users globally. Medisafe's growth is rapidly accelerating as the healthcare industry turns to digital health solutions to support patients. Partnering with top global pharma companies and expanding opportunities across the health ecosystem, Medisafe deploys digital drug companions to holistically support patients throughout their unique therapy journey. Medisafe's advanced technology transforms patient behaviors powered by JITI™ (Justin-Time-Interventions), AI data-driven personalized engine providing support for patients.
Over the past eight years, Medisafe has amassed a database of over four billion dosage behaviors informing JITI to maximize engagement and driving outcomes. In 2020, Medisafe expanded its platform rst launching Medisafe Care Connector which links the all-critical human element into patient support programs digitally connecting patients with clinicians. Last September, Medisafe introduced Maestro, a proprietary no-code technology to build, deploy and optimize personalized journeys. Medisafe Maestro provides a scalable solution to activate patient journeys, orchestrate patient support interventions and increase overall speed to market.
"This investment allows Medisafe to expand holistic treatment support for patients to impact behavior change and ultimately outcomes. Medisafe is continuously advancing its technology to meet the dynamic needs of patients managing complex therapies," said Omri Shor, CEO and Co-Founder of Medisafe. "The future model of patient support is not purely digital but adaptable to empower human connections. In fact, we are seeing impressive results connecting clinicians and care givers into the support solutions with adoption rates 4x that of pure digital and increasing nurse-to-patient connectivity by 1.8x. We are excited to build out further capabilities to meet the evolving needs of the industry."
PRINCETON, N.J. & NEW YORK--(BUSINESS WIRE)--Otsuka Pharmaceutical Development & Commercialization, Inc. (Otsuka) and Click Therapeutics, Inc. announce the initiation of the Mirai study, a landmark fully remote clinical trial to investigate the effectiveness of digital therapeutics in reducing depressive symptoms in adults diagnosed with major depressive disorder (MDD) who are on antidepressant monotherapy.
The pivotal, randomized, controlled trial will enroll up to 540 patients nationwide. Trial participation will be 10 weeks and efficacy will be evaluated as a change from baseline in the Montgomery–Åsberg Depression Rating Scale (MADRS) total score.
“This landmark clinical trial demonstrates Otsuka’s unwavering commitment to the evolution of clinically validated, FDAcleared digital health solutions that support patients living with mental illnesses, like major depressive disorder,” said Kabir Nath, president and CEO, Otsuka North America Pharmaceutical Business Division, Otsuka America, Inc. “The COVID-19 pandemic has clearly demonstrated the need for digital treatments and fully remote e-clinical trials that go beyond the pill to empower patients, enhance connectivity between patients and their healthcare team, and ensure more diverse populations can participate in new clinical trials.”
According to the World Health Organization, depression is a leading cause of disability worldwide, and is a major contributor to the overall global burden of disease. Click and Otsuka are committed to bridging the gaps between the global deficiency of mental health treatment access and the millions of patients in need of safe, effective, convenient and accessible care.
“While awareness of mental illness has grown steadily over the last decade, clinically validated therapeutic options available to patients and providers have remained essentially the same,” said David Benshoof Klein, CEO of Click Therapeutics. “Now more than ever, there is a need for a scalable digital solution that can dramatically expand access to mental health treatment without sacrificing the rigors of clinical validation or a patient-centered focus on engagement and user experience.”
Otsuka and Click Therapeutics Initiate First-of-its-Kind Fully Remote Clinical Trial Using Digital Therapeutics as Adjunctive Therapy in Adults With Major Depressive Disorder Primary objective of the Mirai study is to evaluate the effectiveness of digital therapeutics to reduce depressive symptoms in adults diagnosed with major depressive disorder (MDD) who are on antidepressant monotherapy Trial will run fully remotely utilizing the Verily Project Baseline platform, a first-of-its-kind trial design for MDD Otsuka is committed to exploring digital therapeutics in patients with serious mental illnesses like MDD, which affects more than 17 million adults in the US1 2 / Otsuka and Click will collaborate with Verily, a subsidiary of Alphabet, to execute the trial as a fully remote trial. The collaboration with Verily provides tools and technology to engage patients and clinicians, in order to increase the pace of studies and collect higher quality, more comprehensive data in a more naturalistic setting. The collaboration also enables the trial to proceed efficiently and safely in the face of the unique market challenges presented by COVID-19.
Utrecht, The Netherlands and Philadelphia, USA – February 22, 2021 – LAVA Therapeutics B.V., an early clinical-stage biotechnology company focused on applying its expertise in bispecific gamma-delta T cell engagers to transform cancer therapy, today announced the appointment of Dr. Kapil Dhingra as chairman of its Board of Directors. Dr. Dhingra has more than 30 years of experience in oncology clinical research and drug development within the biotechnology and pharmaceutical industries, as a Board member at several successful oncology biotechnology companies, and as a clinician at academic research centers.
“We are delighted to have Dr. Dhingra join our Board as chairman at this moment in our company’s journey,” said Stephen Hurly, President and Chief Executive Officer. “In the coming year we expect two LAVA programs to enter Phase 1/2a studies in solid and hematologic malignancies. Dr. Dhingra’s deep expertise in cancer drug development and impressive track record of building successful oncology biotechnology companies will have an immediate and beneficial impact on our strategy and mission to bring our first-in-class cancer drugs to patients.”
Mr. Hurly added, “We look forward to benefiting from Dr. Dhingra’s expertise as we expand our preclinical pipeline. Our platform’s modularity enables rapid discovery and development of novel candidates, and Dr. Dhingra’s experience in translational medicine will provide important insights as we select high-value, innovative targets. I also want to thank Erik van den Berg for serving as chairman of LAVA’s Board of Directors since our founding. During his tenure, LAVA raised over $100M in capital from leading life science VCs which provides a solid foundation for the company’s continued growth.”
“I am excited to welcome Dr. Dhingra to the LAVA Board of Directors. His experience will be extremely valuable to LAVA as it transitions to become a clinical-stage company this year,” commented Erik van den Berg.
“LAVA’s novel T cell engager platform, which harnesses the unique attributes of gamma-delta T cells, has the potential to change the standard of care for many high unmet need cancer patient populations,” said Dr. Dhingra. “I look forward to working with the LAVA Board and management team. I also want to thank Erik van den Berg for his leadership of LAVA’s Board. He has been instrumental in growing LAVA into a company with two drug candidates poised to enter the clinic within a year.”
Dr. Dhingra is a medical oncologist and a physician-scientist with a proven track record in academic research, patient care, and drug development. He served as Vice President, Head of the Oncology Disease Biology Leadership Team and Head of Oncology Clinical Development at Hoffmann-La Roche (“Roche”), during which he led numerous drug approvals, including Herceptin®, Tarceva®, and Avastin®. Prior to joining Roche, he worked in the oncology clinical development group at Eli Lilly and Company. Dr. Dhingra has served as a faculty member at The University of Texas M.D. Anderson Cancer Center, Indiana University School of Medicine, and Memorial Sloan Kettering Cancer Center. Dr. Dhingra is currently a member of the Boards of Directors of Black Diamond Therapeutics, Inc., Replimune, Inc., Five Prime Therapeutics, Inc., Autolus Therapeutics plc, and Median Technologies, and he has previously served on the boards of several successful biotech companies, including Biovex, Micromet, Algeta, YM Biosciences, Epitherapeutics, Advanced Accelerator Applications, and Exosome Diagnostics. He is a member of the NCI Experimental Therapeutics Panel. Dr. Dhingra founded KAPital Consulting, LLC in 2008, a company dedicated to helping biotechnology, pharmaceutical, and diagnostic companies realize the www.LavaTherapeutics.com clinical and commercial advances in oncology. Dr. Dhingra obtained his M.B.B.S. degree from the All India Institute of Medical Sciences in New Delhi, India. He completed his residency in internal medicine at Lincoln Medical and Mental Health Center, New York Medical College and completed his fellowship in hematology and oncology at Emory University School of Medicine.
Partnership to create new treatment delivery options for people facing serious diseases
BOSTON, MA—February 10, 2021—i2O Therapeutics, developers of a platform for oral delivery of traditionally injectable biological drugs, announced today a research collaboration with Sanofi to investigate the oral delivery of Sanofi's Nanobody®-based medicines, which are currently administered through intravenous or subcutaneous injections.
Nanobodies - proprietary therapeutic proteins based on camelid derived immunoglobulin single variable domains - have potential uses in the treatment of a range of serious and life-threatening diseases and are being developed in many therapeutic areas including inflammation, hematology, immuno-oncology, oncology and rare diseases. The research collaboration between i2O Therapeutics and Sanofi will explore a new oral route of administering nanobodies.
“Our mission at i2O Therapeutics is to develop safe and effective oral formulations of therapies traditionally limited to injections and we are excited to partner with Sanofi to advance this mission,” said Ravi Srinivasan, co-founder and director of i2O Therapeutics.
“i2O’s ionic liquid platform opens new opportunities to orally deliver biologics, and nanobodies represent an exciting application of this platform,” said Samir Mitragotri, co-founder of i2O Therapeutics.
i2O Therapeutics announced seed funding in April 2020, which was led by Sanofi Ventures, the corporate venture capital arm of Sanofi, and JDRF T1D Fund. The company also announced a strategic investment from Colorcon Ventures, the corporate venture capital fund of Colorcon, Inc. in December 2020.
About i2O Therapeutics
i2O Therapeutics is a biotechnology company developing safe and effective oral formulations of therapies traditionally limited to injections. Using an innovative ionic liquid technology, this platform leverages the benefits of protecting the drug cargo while also transiently enhancing permeation across the epithelial lining when administered orally. i2O is focused on creating the next generation of oral peptide and protein-based therapies. Visit us at www.i2OBio.com.
About Sanofi
Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions. With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe.
Contact
Lauren Arnold
MacDougall
larnold@macbiocom.com
781-235-3060
Strengthens senior leadership team with appointments of Donald Johns, M.D.,
as Chief Medical Officer and Katina Dorton, J.D., MBA, as Chief Financial Officer
CAMBRIDGE, UK, BOSTON and SEATTLE – December 15, 2020 – NodThera, a biotechnology company developing a new class of medicines that inhibit the NLRP3 inflammasome to treat chronic inflammation, today announced the expansion of its senior leadership team with the appointments of Donald Johns, M.D., as Chief Medical Officer and Katina Dorton, J.D., MBA, as Chief Financial Officer.
Dr. Johns is an accomplished drug development leader and board-certified clinical neurologist who previously served as Chief Medical Officer and Executive Vice President of Medical and Scientific Affairs at Syntimmune, prior to the company’s acquisition by Alexion Pharmaceuticals. Ms. Dorton is a recognized and internationally experienced financial executive, corporate director and public company CFO. Ms. Dorton’s industry expertise includes roles in healthcare, life sciences and investment banking, including experiences at Repare Therapeutics and AVROBIO, Inc.
NodThera is advancing a portfolio of potent and selective inhibitors of the NLRP3 inflammasome that reduce both IL-1β and IL-18, pro-inflammatory cytokines which are known to play a role in chronic inflammation underlying a wide range of diseases. The pipeline includes brain penetrant NLRP3 inhibitors for central nervous system (CNS) indications.
“We welcome Don and Katina to the team as we continue to advance our portfolio of differentiated NLRP3 inhibitors to capitalize on the opportunity to exploit the well-understood, but still untapped therapeutic potential of the NLRP3 inflammasome across a broad spectrum of diseases,” said Adam Keeney, Ph.D., President & Chief Executive Officer of NodThera. “Don is an accomplished clinician with an impressive track record who brings a wealth of innovative clinical development experience. Katina brings capital markets experience and has built financial, legal and operational functions to support companies through aggressive growth, including IPO preparation. Their combined contributions will add significant value as we continue to advance NodThera’s portfolio through clinical development and additional financing rounds.”
“The NLRP3 inflammasome is one of the most exciting emerging areas of therapeutic science. NodThera’s best-in-class molecules have great potential to address the unmet medical need in patients with a wide range of inflammatory disorders,” said Dr. Johns. Ms. Dorton adds, “By leveraging innate immunity, NodThera’s platform has the potential to blaze a new path forward in a wide range of disorders, and I am excited to join the rest of the experienced leadership team in this effort.”
Dr. Johns brings more than 25 years of experience in the development of novel treatments for serious diseases, including autoimmune and CNS disorders. Prior to his role at Syntimmune, Dr. Johns served in key leadership positions at Biogen and the Novartis Institutes for Biomedical Research (NIBR). He has contributed to numerous investigational new drug applications (INDs), first-in-human studies and conclusive proof-of-concept studies in a broad spectrum of CNS and autoimmune diseases, as well as four successful new drug applications (NDAs). Prior to joining the pharmaceutical industry, he was a neurologist clinician-scientist at Johns Hopkins and Harvard Medical School. Dr. Johns earned his B.A. from Vanderbilt University and his M.D. from the Yale University School of Medicine.
Ms. Dorton currently serves on the board of directors for Fulcrum Therapeutics (Nasdaq: FULC), Pandion Therapeutics, Inc. (Nasdaq: PAND) and US Ecology (Nasdaq: ECOL). She most recently served as Chief Financial Officer of Repare Therapeutics, a synthetic lethality and DNA repair-focused oncology company. Prior to Repare, Ms. Dorton served as Chief Financial Officer of AVROBIO, a lentiviral gene therapy company. Earlier in her career, she served as a managing director in investment banking for Morgan Stanley and Needham & Company and as an associate attorney at Sullivan & Cromwell. Ms. Dorton received her J.D. from the University of Virginia School of Law, her MBA from George Washington University and her B.A. from Duke University.
About NodThera
NodThera is a biotechnology company developing a new class of potent and selective NLRP3 inflammasome inhibitors for the treatment of diseases driven by chronic inflammation. Led by an experienced management team, NodThera is leveraging new insights into inflammasome biology and chemistry to build a platform of highly differentiated small molecule NLRP3 inhibitors. The company was founded by Epidarex Capital and further financed by 5AM Ventures, Cowen Healthcare Investments, F-Prime Capital, Novo Holdings, Sanofi Ventures and Sofinnova Partners. NodThera was founded in 2016 and maintains offices in Cambridge, UK, Seattle, WA and Boston, MA. For more information please visit www.nodthera.com.
Media Contact
Gina Nugent
Ten Bridge Communications
(617) 460-3579
gina@tenbridgecommunications.com
- On the back of highly promising Phase Ib data, MinervaX has raised financing from leading investors to accelerate development of its novel vaccine through the end of Phase II trials and preparations for Phase III pivotal trials
- Sanofi Ventures, Wellington Partners, Adjuvant Capital, and Industrifonden join existing investors Novo Holdings REPAIR Impact Fund, Sunstone Life Science Ventures and LF Investment
- Group B Streptococcus (GBS) is one of the leading causes of stillbirth and infant mortality representing a significant unmet need globally, including in the US and Europe; nearly one in five women globally are colonized by GBS
Copenhagen, Denmark, 15 December 2020 – MinervaX, a privately held Danish biotechnology company developing a novel vaccine against Group B Streptococcus (GBS), announced today that it has raised an upsized EUR 47.4 million Series B financing. The round included new investors Sanofi Ventures, Wellington Partners, Adjuvant Capital, and Industrifonden, along with existing investors Novo Holdings REPAIR Impact Fund, Sunstone Life Science Ventures, and LF Investment. Proceeds will advance the clinical development of MinervaX’s novel GBS vaccine through Phase II clinical trials, as well as manufacturing and regulatory preparation for Phase III.
Concurrent with the financing, Christopher Gagliardi from Sanofi Ventures, Karl Nägler from Wellington Partners, Kabeer Aziz from Adjuvant Capital and Bita Sehat from Industrifonden will join MinervaX’s board of directors.
GBS is responsible for nearly half of all life-threatening infections in newborns. MinervaX’s protein-only GBS vaccine targets pregnant women for the prevention of adverse pregnancy outcomes and life-threatening neonatal infections associated with GBS. Globally, 15-25% of women are colonized with GBS, and they run the risk of transmitting the bacteria to their child in utero, during birth and / or during their first months of life. GBS colonization may lead to late-term abortions, premature delivery or stillbirth; and in newborn children may result in sepsis, pneumonia or meningitis, all of which carry a significant risk of severe morbidity, long-term disability or death.
Currently, the only preventative strategy available involves the use of intravenously delivered prophylactic antibiotics, which does not comprehensively prevent GBS infection in utero or protect against late-onset infection in newborns. As this approach is expensive and logistically challenging, it fails to cover all, including the most severe cases in the US and Europe, nor is it available in resource-limited settings.
Commenting on the financing, Per Fischer, CEO of MinervaX, said: “Prevention of GBS infections in pregnant women and newborns represents a large unmet medical need. The current preventive strategy is insufficient and involves excessive use of prophylactic antibiotics, which has resulted in the emergence of wide-spread antibiotic resistance.
“We are pleased to have received funding from such a strong investment syndicate. It is a significant endorsement of the potential of our vaccine. We look forward to advancing our novel vaccine candidate through Phase II clinical trials to develop a new standard of care in preventing GBS infections.”
Commenting on the investment, Christopher Gagliardi, Director of Investments at Sanofi Ventures, said: “Sanofi Ventures is tremendously excited by the first and best in class potential of MinervaX’s GBS vaccine. We are thrilled to invest alongside a top-tier investor syndicate while supporting Sanofi’s strategic goals and commitment to early stage companies advancing global public health.”
Karl Nägler, Managing Partner at Wellington Partners said: “We are proud and excited to back MinervaX’s GBS program that will address an unmet high medical need and represents a blockbuster commercial opportunity. Beyond prevention of GBS infections in newborns, we are eager to explore important further indications for this much needed vaccine.”
Emmanuelle Coutanceau, Partner at Novo Seeds and Board Member at MinervaX, added: “MinervaX is developing an important vaccine against a potentially fatal pathogen and, in doing so, is furthering the battle against antimicrobial resistance. This is a landmark for the Novo Holdings REPAIR Impact Fund with the first company in the fund moving to Phase II. We are also delighted to help bring together such a strong syndicate in a company where Novo was one of the first investors.”
MinervaX has completed Phase I studies across 300 healthy female subjects, generating compelling data to support advancing its novel vaccine candidate to Phase II trials. Studies to date have demonstrated a favourable safety profile, while generating high levels of long-lasting antibodies, which are capable of mobilizing the immune system against GBS bacteria and preventing invasion of epithelial and endothelial cell barriers.
The development of MinervaX’s novel GBS vaccine candidate is also endorsed by Group B Strep Support and Group B Strep International, and GBS has been prioritised by a number of public health organisations. Both increased uptake of immunisation among pregnant women and greater awareness of the implications of GBS suggest that a safe and effective vaccine targeting GBS would be well suited to address this unmet need.
Enquiries
For more information on MinervaX, please contact:
Optimum Strategic Communications
Supriya Mathur, Charlotte Hepburne-Scott
+44 (0) 203 922 0891, +44 (0) 787 687 2223
minervax@optimumcomms.com
About MinervaX
MinervaX is a Danish biotechnology company, established in 2010 in order to develop a prophylactic vaccine against Group B Streptococcus (GBS), based on research from Lund University. MinervaX is developing a GBS vaccine for maternal immunization, likely to have superior characteristics compared with other GBS vaccine candidates in development. The latter are based on traditional capsular polysaccharide (CPS) conjugate technology. By contrast, MinervaX’s vaccine is a protein-only vaccine based on fusions of highly immunogenic and protective protein domains from selected surface proteins of GBS (the Alpha-like protein family). Given the broad distribution of proteins contained in the vaccine on GBS strains globally, it is expected that MinervaX’s vaccine will confer protection against virtually 100% of all GBS isolates. www.minervax.com
About Group B Streptococcus (GBS)
GBS is responsible for nearly 50% of all life-threatening infections in newborns. At any given time, some 15-25% of women are spontaneously colonized with GBS, and they run the risk of transmitting the bacteria to their child in the womb, during birth and/or during the first months of life. GBS colonization may lead to late abortions, premature delivery or stillbirth and, in the newborn child, may result in sepsis, pneumonia or meningitis, all of which carry a significant risk of severe morbidity, long-term disability or death.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi. Sanofi Ventures invests in early-stage biotech and digital health companies with innovative ideas and transformative new products and technologies of strategic interest to Sanofi. Among these areas are vaccines, oncology, immunology, rare diseases, potential cures in other core areas of Sanofi’s business footprint, and digital health solutions. For more information, visit www.sanofiventures.com.
About Novo Holdings A/S
Novo Holdings is recognized as a leading international life science investor, with a focus on creating long-term value. As a life science investor, Novo Holdings provides seed and venture capital to development-stage companies and takes significant ownership positions in growth and well-established companies. Novo Holdings also manages a broad portfolio of diversified financial assets. Further information: http://www.novoholdings.dk
About REPAIR Impact Fund
The Fund invests in start-ups, early-stage companies and corporate spin-outs around the world. It gives priority to first-in-class therapies, covering small molecules, biologics and new modalities, from the early stage of drug development (lead optimization) to later stages of clinical development (into Phase 2). It can invest as the sole investor or in a syndicate, with investments ranging from EUR 1 million to EUR 12 million.
The projects are selected through an investment process with support from a highly qualified Scientific Selection Board, comprising ten world-class experts. For more information about members of the Scientific Selection Board, see www.repair-impact-fund.com/people.
The Fund focuses on priority pathogens as defined by the World Health Organization and the United States Centers for Disease Control and Prevention, a catalogue of 18 families of bacterial and fungal pathogens that pose the greatest threat to human health. For more details about the investment process, see www.repair-impact-fund.com/investment-process.
REPAIR is an acronym: Replenishing and Enabling the Pipeline for Anti-Infective Resistance
About Wellington Partners
Wellington Partners is a leading European venture capital firm investing in early- and growth-stage life science companies. Wellington Partners is focused on investing in the most promising life science companies in the fields of biotechnology, therapeutics, medical technology, diagnostics and digital health. With funds totaling more than €1 billion, thereof €430 million committed to Life Sciences, Wellington Partners has been actively supporting world class private companies translating true innovation into successful businesses with exceptional growth. To date, Wellington Partners has invested in 46 innovative life science companies, including Actelion (acquired by J&J), Definiens (acquired by AZ), Invendo (acquired by Ambu), Rigontec (acquired by MSD), Symetis (acquired by Boston Scientific), and Themis (acquired by MSD). www.wellington-partners.com
About Adjuvant Capital
Adjuvant is a New York- and San Francisco-based life sciences investment fund built to accelerate the development of new technologies for the world's most pressing public health challenges. Backed by prominent healthcare investors such as Novartis, Merck, the International Finance Corporation, and the Bill & Melinda Gates Foundation, Adjuvant draws upon its global network of scientists, public health experts, biopharmaceutical industry veterans, and development finance professionals to identify new investment opportunities. Adjuvant invests in companies developing promising new vaccines, therapeutics, and diagnostics for historically overlooked indications targeting high-burden infectious diseases, maternal and child health, and antimicrobial resistance, with a commitment to make these interventions accessible to those who need them most in low- and middle-income countries. For more information, visit www.adjuvantcapital.com
About Industrifonden
Industrifonden is a Nordic venture capital investor based in Stockholm that invests in early-stage growth companies. Our areas of expertise include Life Sciences, Deep Tech and Transformative Tech. In the life science space, our focus is on biotech, heathtech and medtech, and our life-science portfolio includes companies like Oncopeptides, Calliditas and Bonesuppport. www.industrifonden.com
About Sunstone Life Science Ventures
Sunstone Life Science Ventures is an independent European venture capital investment firm founded in 2007 by an international team of industry experts with combined entrepreneurial, operational and financial experience. Sunstone Life Science Ventures focuses on developing and expanding early-stage Life Science companies with strong potential to achieve global success in their markets. Since the inception, Sunstone Life Science Ventures has invested in more than 50 companies in the areas of pharmaceuticals, medical technologies and diagnostics, and has completed more than 20 successful IPOs and large M&A transactions. Managing total funds of approx. €500 million, Sunstone Life Science Ventures is one of the largest Nordic venture capital investors. https://sunstone.eu/
LF Investment
LF Investment is an investment company fully owned by The Lauritzen Foundation. www.lauritzenfonden.com
Preclinical data published in Cell show the company’s COVID-19 vaccine candidate induces high neutralizing antibody titers after a single administration
SEATTLE, October 30, 2020 – Icosavax, Inc. today announced the launch of the company’s COVID-19 vaccine program with preclinical data on the company’s VLP vaccine candidate, IVX-411, which comprises a virus-like particle (VLP) displaying the SARS-CoV-2 receptor-binding domain (RBD) in a highly immunogenic array. Icosavax also announced that the Bill & Melinda Gates Foundation has provided a $10 million grant to support the company’s COVID-19 vaccine program through the first in human Phase 1 clinical trial in young and older adults, expected to initiate in mid-2021. In addition, Icosavax received $6.5 million from Open Philanthropy to support development of the company’s vaccine platform technology and COVID-19 vaccine candidate. The company is currently advancing the necessary studies to support regulatory filings and has initiated GMP manufacturing. To enable rapid progress of the company’s COVID-19 vaccine candidate to the clinic, Amgen has agreed to manufacture a key intermediate for initial clinical studies.
“This is truly a great example of the scientific community coming together in a time of exceptional need to fight this pandemic. We are grateful to the Bill & Melinda Gates Foundation and Open Philanthropy for their financial support and to Amgen for the supply of a key intermediate for manufacturing,” said Adam Simpson, Chief Executive Officer of Icosavax. “The team at Icosavax is dedicated to advancing vaccines against severe life-threatening respiratory diseases to protect our most at-risk populations. Because VLP vaccines have the potential to induce high-neutralizing antibody titers, our COVID-19 VLP vaccine candidate could be especially important for older adults with age-related declines in immunity.”
Developed by scientists at the University of Washington School of Medicine using structure-based vaccine design techniques invented at the Institute for Protein Design (IPD) at the UW Medicine, IVX-411, the lead vaccine candidate for COVID-19, is a self-assembling protein nanoparticle that displays 60 copies of the SARSCoV- 2 spike (S) glycoprotein receptor-binding domain in a highly immunogenic array. Preclinical data from UW researchers and their collaborators show IVX-411 induces high neutralizing antibody titers in mice after a single administration and further improvement after a second administration (Cell 2020). Titers after the second administration were ten-fold higher than those seen with the soluble SARS-CoV-2 spike protein that forms the basis of many other vaccine candidates. The data also show a strong B-cell response after immunization, critical for immune memory and a durable vaccine effect, with antibodies that target multiple distinct epitopes on the RBD, suggesting potential protection from escape mutations.
“These data highlight that while our VLP-based SARS-CoV-2 vaccine may not be first to market, it has the potential to be a best-in-class vaccine that safely delivers potent and durable immune protection,” said Icosavax co-founder Neil King, Ph.D. Dr. King is also inventor of the computationally designed VLP technology at the Institute for Protein Design, and assistant professor of biochemistry at the UW School of Medicine. “This technology is designed to drive higher neutralizing antibody titers than soluble protein approaches and to create safe, stable, and effective VLP vaccines with simple and scalable manufacturing.”
“It’s clear that we will need a variety of vaccine approaches to effectively fight this novel coronavirus,” said Jean-Paul Prieels, Ph.D., former Senior Vice President of Research and Development at GSK Vaccines and member of the scientific advisory board of Icosavax. “That’s why it’s important to advance not just the fastest but the best vaccine technologies that can deliver safe, effective, and durable protection.”
VLPs enable high-density, multivalent display of antigens in a manner that closely resembles viruses, with an important difference. VLPs contain no genetic material, so they are non-infectious and can provide a safer alternative to live-attenuated or inactivated vaccines. The high yield and stability of the protein components and assembled nanoparticles suggest that manufacture of the nanoparticle vaccines will be highly scalable.
Icosavax has a worldwide license with an exclusive option for IVX-411 in North America and Europe from the University of Washington.
About Icosavax
Icosavax is focused on developing safe and effective vaccines against infectious diseases that cause severe, life-threatening respiratory illnesses. In addition to the COVID-19 vaccine candidate, Icosavax is also advancing IVX-121 into clinical trials as a potential vaccine for respiratory syncytial virus (RSV) for older adults. The company was founded on breakthrough computationally-designed virus like particle technology developed at the Institute for Protein Design and exclusively licensed from the University of Washington. Icosavax is located in Seattle. For more information, please visit www.icosavax.com.
Media Contact:
Jessica Yingling, Ph.D.,
Little Dog Communications Inc.,
jessica@litldog.com,
+1.858.344.8091
Financing round led by Novo Ventures and includes new investors Cowen Healthcare
Investments and Sanofi Ventures, as well as existing investors 5AM Ventures, F-Prime Capital,
Sofinnova Partners and founding investor Epidarex Capital
Proceeds to support advancement of pipeline of small molecule NLRP3 inflammasome inhibitors,
including continued progression of NT-0167 through clinical development
CAMBRIDGE, UK, BOSTON & SEATTLE – June 3, 2020 – NodThera, a clinical stage biotechnology company developing a new class of medicines that inhibit the NLRP3 inflammasome to treat diseases driven by chronic inflammation, today announced that it has secured $55 million (£44 million) in a Series B financing. NodThera’s lead candidate, NT-0167, is being evaluated in a Phase 1 clinical trial in healthy volunteers.
“Millions of people live with diseases in which chronic inflammation plays a role,” said Nanna Lüneborg, Partner, Novo Ventures. “NodThera’s approach to selectively target the NLRP3 inflammasome without broadly suppressing the immune system is an enormously promising strategy for developing therapies that can help these patients.”
Proceeds from the financing will be used to advance NT-0167 through clinical studies, further progress the development of additional compounds — including brain-penetrant NLRP3 inflammasome inhibitors for central nervous system indications — and continued drug discovery efforts.
“The NLRP3 inflammasome is one of the most exciting emerging areas of drug discovery. Therapeutics that disrupt the NLRP3 inflammasome to inhibit damaging inflammatory processes have the potential to help patients with many chronic diseases,” said Henrijette Richter, Managing Partner at Sofinnova Partners. “NodThera has distinguished their science in a field that holds huge promise and we look forward to the company’s further progress.”
The first-in-human clinical study for NT-0167 is evaluating the candidate for safety and tolerability as well as pharmacokinetic and pharmacodynamic response. In preclinical studies, NT-0167 demonstrated potent and selective inhibition of the NLRP3 inflammasome resulting in reductions of IL-1β and IL-18, pro-inflammatory cytokines which are known to play a key role in chronic inflammation underlying a wide range of diseases.
“Continued advancement of our lead molecule through clinical development represents a significant achievement in our work to exploit the still untapped therapeutic potential of selectively targeting the NLRP3 inflammasome,” said Adam Keeney, President and Chief
Executive Officer of NodThera. “This financing from a world-class syndicate of life science investors speaks to the significant potential of our pipeline.”
Joining the NodThera Board as part of this Series B financing will be Nanna Lüneborg, Partner at Novo Ventures, Kevin Raidy, Managing Partner at Cowen Healthcare Investments and Alex Pasteur, Partner at F-Prime Capital.
About NodThera
NodThera is a biotechnology company developing a new class of potent and selective NLRP3 inflammasome inhibitors for the treatment of diseases driven by chronic inflammation. Led by an experienced management team, NodThera is leveraging new insights into inflammasome biology and chemistry to build a platform of highly differentiated small molecule NLRP3 inhibitors. The company was founded by Epidarex Capital and further financed by 5AM Ventures, Cowen Healthcare Investments, F-Prime Capital, Novo Ventures, Sanofi Ventures and Sofinnova Partners. NodThera maintains offices in Cambridge, UK, Seattle, WA and Boston, MA.
For more information please visit www.nodthera.com.
Media Contact
Matthew Corcoran
Ten Bridge Communications
(617) 866-7350
mcorcoran@tenbridgecommunications.com

- BIAL Biotech to be based in Cambridge, Massachusetts and will be a Research Center of Excellence dedicated to genetically-defined Parkinson’s disease
- LTI-291 clinical program and other research programs in Parkinson's disease acquired from Lysosomal Therapeutics, Inc
- R&D team led by Peter Lansbury, professor of neurology at Harvard Medical School
- Investment may add up to 130 million dollars depending on the accomplishment of downstream development, and several regulatory and commercial milestones
Porto / Cambridge, Mass, October 1, 2020 – BIAL, a pharmaceutical company based in Portugal with locations across Europe and dedicated to R&D in CNS diseases, announced today that it has established a new affiliate in the United States of America, BIAL Biotech Investments Inc. (BIAL Biotech). This new research center focused on genetically-defined Parkinson’s disease is based in Cambridge, Massachusetts, a prominent biotech hub in the world.
Simultaneously, BIAL announced that it has acquired worldwide rights of LTI-291 and all the Parkinson’s disease research programs of Lysosomal Therapeutics Inc. (LTI) and taken on the entire R&D team.
António Portela, executive president of BIAL, reveals “Our entry into the US with the creation of BIAL Biotech and the acquisition of the promising programs from LTI, is a decisive step towards the fulfilment of our mission to contribute to improving the quality of life of people worldwide. The development of this new research center in the US, is a landmark of enormous relevance for us. We are investing in science and research, through our direct presence in one of the most important research hubs in the world and in one of the most promising areas of medicine”.
This acquisition not only provides the company with a pipeline of new product candidates in Parkinson’s disease but also an experienced R&D team, led by Peter Lansbury, professor of neurology at Harvard Medical School and a recognized thought leader in the field of neurodegenerative diseases.
With this acquisition, BIAL is expanding its pipeline, namely with the integration of new compounds in neurodegeneration already in clinical development, specifically for Parkinson's disease, where the pharmaceutical company already has a significant market position.
The executive president of BIAL also points out: “The compounds we’ve acquired are based on genetics, a new field of research for us. The lead asset, which now has the code name 'BIA 28-6156/LTI-291', has an innovative mechanism of action and presents the potential of being a first disease-modifying therapy for a genetic subset of Parkinson’s disease. It has successfully completed a Phase I trial program and should be ready to start Phase II studies in 2021. We´re progressing from symptomatic treatment to an intervention in the mechanisms ase, which is very exciting for BIAL”.
“We are happy to be part of BIAL and take the lead on the growth story in the US” says Kees Been, former CEO of LTI and now CEO of BIAL Biotech. "With the commitment and resources of BIAL we will be able to accelerate our novel clinical and research programs as targeted and personalized treatment approaches for genetically-defined PD patients”.
“On behalf of the LTI shareholders, I want to express our enthusiastic endorsement of BIAL as a good home for the programs of LTI”, said Clay B. Thorp, General Partner, Hatteras Venture Partners, “This deal is a perfect fit and we have great confidence that the team, with the resources and commitment of BIAL, will advance LTI-291 and the other compounds rapidly to patients to treat this debilitating form of Parkinson’s disease.”
BIA 28-6156 / LTI-291 is a drug compound aimed at treating patients with GBA-Associated Parkinson’s diseases (GBA-PD) caused by an underlying mutation in the GBA1 gene. This genetic mutation affects approximately 10% to 15% of Parkinson’s disease patients (100,000-150,000 patients in the US) and is a validated risk factor that generally leads to an earlier age of disease onset and more rapid disease progression.
In addition to an upfront payment, the total value of the deal could reach up to 130 million dollars, based on attainment of a series of milestone payments as the assets progress through the clinic and into the market. Other financial terms will not be disclosed.
About BIAL Biotech
BIAL Biotech is a Center of Excellence for the development of genetically-defined Parkinson’s disease therapeutics located in the heart of the Boston biotech cluster. BIAL Biotech is a wholly owned subsidiary of BIAL, a pharmaceutical company based in Portugal with locations across Europe.
About BIAL
Founded in 1924, BIAL's mission is to research, develop and provide therapeutic solutions within the area of health. In the last decades, BIAL has focused strategically on quality, innovation and internationalisation. BIAL is strongly committed to therapeutic innovation, investing more than 20% of its annual turnover into research and development within neurosciences and the cardiovascular system. The company expects to introduce new drugs on the market in the coming years, strengthening its international presence based on proprietary drugs and achieving its goal of supplying innovative products to patients worldwide.
For more information on BIAL: www.bial.com
Media Contacts:
BIAL
Susana Vasconcelos
susana.vasconcelos@bial.com
US Media Contact:
Amanda Whelan
MacDougall Advisors
awhelan@macbiocom.com
Funding to support advancement of powerful bispecific gamma-delta T cell engager programs
for the treatment of a broad range of cancers
New investors include Novo Holdings, Sanofi Ventures, Redmile, Ysios and BB Pureos
Utrecht, The Netherlands and Philadelphia, USA – September 17, 2020 – LAVA Therapeutics, a biotech company pioneering the development of bispecific antibodies to engage gamma-delta T cells for cancer therapies, today announced the closing of an oversubscribed $83 million (€71 million) Series C financing to fund the advancement of its pipeline and platform. The financing was co-led by new investors Novo Ventures, the venture arm of Novo Holdings, and Sanofi Ventures, and included additional new investors Redmile Group, LLC, Ysios Capital and BB Pureos Bioventures. In addition, current investors Versant, Gilde Healthcare and MRL Ventures Fund, LLC participated significantly in the round.
As part of the transaction, Nanna Lüneborg, partner at Novo Ventures, Laia Crespo, EU head of investments at Sanofi Ventures, and Joël Jean-Mairet, managing partner and co-founder of Ysios, joined LAVA as members of the board of directors.
“We are grateful to have attracted a high-quality syndicate of new investors complementing strong continued support of our existing investors. This financing provides meaningful capital to advance our bispecific gamma-delta T cell engager portfolio into multiple proof-of-concept clinical trials expected to start in 2021 for the treatment of solid tumors and hematologic malignancies,” said Stephen Hurly, chief executive officer of LAVA Therapeutics. “We believe our targeted approach, leveraging the unique features of gamma9-delta2 T cells with innovative bispecific antibodies, will deliver novel T cell-based therapies offering advantages over today’s oncology treatments. In addition to the funding raised, the appointments of Nanna, Laia and Joël to our board further strengthen our team, and we look forward to benefiting from their insights and industry expertise.”
Gamma-delta T cells are the natural surveillance cells of the immune system, continuously patrolling the human body for the identification and targeting of tumor cells. These cells bridge the innate with the adaptive immune system and are a largely untapped opportunity in cancer treatment. LAVA’s bispecific gamma-delta T cell engager platform is harnessing the unique properties of these T cells creating a revolutionary truly tumor-targeted immunotherapy to improve outcomes for cancer patients.
“LAVA’s bispecific antibody approach to targeting and engaging gamma-delta T cells has the potential to transform the treatment of a wide range of cancers,” said Dr. Lüneborg. “We are impressed by the preclinical data generated by LAVA to date, which validate the company’s platform and support their transition into a clinical-stage organization. The team is highly experienced in drug development, and I look forward to working with them.”
“Gamma-delta T cells are an emerging field and an incredibly exciting area in oncology. Bispecific antibodies able to directly engage this type of cells offer the potential to significantly impact patients across the globe in these diseases with high morbidity and mortality,” said Dr. Crespo.
About LAVA Therapeutics
LAVA Therapeutics is developing a proprietary bispecific antibody platform that engages gamma-delta T cells for the treatment of hematological and solid cancers. The company’s firstin- class immuno-oncology approach activates Vγ9Vδ2 T cells upon binding to membraneexpressed tumor targets. LAVA was founded in 2016 based on intellectual property originating from the Amsterdam University Medical Center. The company has established a highly experienced antibody research and development team located in Utrecht, the Netherlands (headquarters) and Philadelphia, USA. For more information, please visit www.lavatherapeutics.com.
Contact
Alicia Davis
THRUST Strategic Communications
alicia@thrustsc.com
- Proceeds support clinical development for lead program in cholestatic and uremic pruritis and second program for mast cell-related diseases
- Financing round led by Sanofi Ventures and Cowen Healthcare Investments and included new investors Redmile Group and Perceptive Advisors
- Jason Hafler, Ph.D., of Sanofi Ventures and Kevin Raidy of Cowen Healthcare Investments join Escient board of directors
San Diego, CA – September 14, 2020 – Escient Pharmaceuticals, Inc., an industry-leading company developing Mas-related G Protein-Coupled Receptor (MRGPR)-targeted drugs to address serious, underserved medical needs across a broad range of therapeutic indications, today announced the completion of a $77.5 million Series B financing and the initiation of a Phase 1/1b clinical trial of EP547, a MRGPRX4-targeted product candidate to treat cholestatic and uremic pruritis. The round was led by Sanofi Ventures and Cowen Healthcare Investments (CHI) with participation by new investors Redmile Group and Perceptive Advisors. All of the company’s existing investors, including The Column Group, 5AM Ventures, and Osage University Partners, participated in the round. Escient discovered and is developing EP547 based on the novel finding of disease-related accumulation of key metabolites that specifically activate the MRGPRX4 itch receptor and will use these proceeds to support clinical development. The company is developing a portfolio of MRGPR-focused therapeutics including a second program targeting mast cell-based MRGPRX2 that is anticipated to play a role in a number of indications in various therapeutic areas.
“In just over two years since our launch, we’ve made significant progress developing our platform and pipeline. We have advanced both our understanding of the role MRGPRs in a number of therapeutic areas and our methods for drugging them to potentially treat several diseases based on novel and specific mechanisms of action,” said Alain Baron, M.D., Chief Executive Officer of Escient. “Through our work, we’ve identified promising drug candidates that we are advancing into clinical development with the potential to build a pipeline of class-leading, once-daily, oral therapeutics to address debilitating diseases.”
Effective treatments for severe pruritus associated with cholestasis and uremia are sorely needed. Drug development to address pruritus in these conditions has been hampered by the lack of basic biological knowledge of their specific mechanisms. Cholestasis occurs in chronic adult and pediatric conditions wherein 30-100% of patients experience moderate-to-severe itch. Hemodialysis and end-stage renal disease are largely adult conditions with up to 55% of the population experiencing itch.
“Current treatment options for cholestatic and uremic pruritis are non-specific, largely ineffective, and have significant side effects,” said Marcus F. Boehm, Ph.D., Chief Scientific Officer of Escient. “Based on Escient Series B Financing/Page 2 our preclinical discoveries, we believe we have the opportunity to develop a mechanism-based, well- tolerated, rapidly-acting and highly effective novel therapy for these patients with the potential to make a significant difference in their lives.”
The Phase 1/1b trial of EP547 is a randomized, double-blind, placebo-controlled study to evaluate the safety, tolerability, and pharmacokinetics of single and multiple ascending doses of EP547 in healthy subjects and subjects with cholestatic or uremic pruritus.
In conjunction with the financing, Jason Hafler, Ph.D., of Sanofi Ventures and Kevin Raidy of CHI have joined Escient’s board of directors.
About Escient Pharmaceuticals
Escient Pharmaceuticals is a biotechnology company that is advancing first-in-class G Protein-Coupled Receptor (GPCR)-targeted drugs to address serious, unserved medical needs across a broad range of therapeutic indications. Focused on unleashing the therapeutic potential of specific orphan GPCRs, including the novel family of Mas-Related G-Protein Receptors (MRGPRs), Escient’s lead program targets MRGPRX4. Based in San Diego, Calif., Escient is led by an experienced team of biotechnology entrepreneurs with specific expertise in GPCR drug discovery and development, and funded by top-tier life science investors, including The Column Group, 5AM Ventures, Osage University Partners and Altitude Life Science Ventures. Visit www.escientpharma.com to learn more.
Contact:
Cory Tromblee
Scient Public Relations
cory@scientpr.com
William Hodder
Escient Pharmaceuticals, Inc.
info@escientpharma.com
In a significantly oversubscribed offering, Science 37 attracted capital to support its rapid growth, expand its technology platform and accelerate global expansion.
LOS ANGELES, August 20, 2020 — Science 37, the industry leader in decentralized clinical trials, today announced it closed an oversubscribed $40 million funding round. Existing investors Lux Capital, Redmile Group, and PPD, Inc. (Nasdaq: PPD) led the round, and are joined by existing investors Novartis, Amgen, Sanofi Ventures, GV, and Glynn Capital. Notable new investors include LifeSci Ventures and Mubadala Ventures.
Science 37 will use the new capital to support its rapid growth, expand its technology platform, and accelerate its global expansion efforts—further strengthening its ability to help sponsors execute decentralized trials and enable patients to participate in trials from anywhere, without the burden of traveling to a traditional clinical site.
“The Science 37 mission to democratize clinical research by bringing trials directly to the patients is not only the right thing to do, but it also has had a clear, measurable impact on sponsors’ ability to access a more diverse patient population, accelerate research, and bring new medicines to the market faster,” said David Coman, CEO of Science 37. “We are thrilled to have an incredibly strong, committed network of investors and plan to continue extending our leadership in the decentralized clinical trial market.”
The new investment builds upon immense progress from the company’s previous round, which afforded global expansion; a progressive, BYOD patient experience; world-class collaboration workflow; a unified regional nurse network; and advanced patient recruitment capability. The company also embedded clinical industry veterans in management and implemented a flexible business model to enable a choice between fully decentralized trials, a Science 37 Metasite™—a virtual arm to a traditional site network—and the ability to purchase Science 37 technology with optional services.
“We are in the unique position where the clinical trial industry absolutely has to adapt to a new model, today and going forward,” says Adam Goulburn, partner at Lux Capital. “Science 37 has been preparing for this day for years with an incredible head start. They are uniquely positioned to help sponsors conduct clinical research without the traditional barriers. With their foundation and impressive growth, Lux knows Science 37 will continue to lead the way.”
About Science 37
Science 37 is making the promise of virtual trials the new reality. By engaging with patients from the comfort of their own home, we provide access to patients who can never be reached by traditional site-based models. We have proven to enroll faster, retain patients at a higher rate, and reach a more representative population. Science 37 has conducted more decentralized, interventional trials than any other company, using an expansive network of telemedicine investigators and home-health nurses, who are supported by the industry’s most comprehensive, fully integrated, decentralized clinical trial platform. Learn more at Science 37, and follow Science 37 on Twitter, LinkedIn, and Facebook.
Contact:
Lawrence Lloyd Science 37
Phone: (984) 377-3737
Email: pr@science37.com
# # #
WATERTOWN, Mass., Aug. 20, 2020 (GLOBE NEWSWIRE) -- Kymera Therapeutics, Inc. (NASDAQ: KYMR), a biopharmaceutical company focused on discovering and developing novel small molecule therapeutics that selectively degrade disease-causing proteins by harnessing the body’s own natural protein degradation system, today announced the pricing of its upsized initial public offering of 8,684,800 common shares at a public offering price of $20.00 per share. All of the shares are being offered by Kymera. The gross proceeds of the offering, before deducting underwriting discounts and commissions, and other offering expenses payable by Kymera, are expected to be approximately $173.7 million. Kymera has granted the underwriters a 30-day option to purchase up to an additional 1,302,720 common shares at the initial public offering price.
The shares are expected to begin trading on the Nasdaq Global Market on August 21, 2020 under the ticker symbol “KYMR.” The offering is expected to close on August 25, 2020, subject to the satisfaction of customary closing conditions.
Morgan Stanley, BofA Securities, Cowen, and Guggenheim Securities, are acting as joint book-running managers for the offering.
A registration statement relating to these securities became effective on August 20, 2020. The offering will be made only by means of a prospectus. Copies of the final prospectus, when available, may be obtained from Morgan Stanley & Co. LLC, Attention: Prospectus Department, 180 Varick Street, 2nd Floor, New York, NY 10014, or by email at prospectus@morganstanley.com; BofA Securities, NC1-004-03-43, 200 North College Street, 3rd floor, Charlotte, NC 28255- 0001 Attn: Prospectus Department, or by email at dg.prospectus_requests@bofa.com; Cowen and Company, LLC, c/o
Broadridge Financial Solutions, 1155 Long Island Avenue, Edgewood, NY, 11717, Attention: Prospectus Department, by telephone at (833) 297-2926; Guggenheim Securities, LLC, Attention: Equity Syndicate Department, 330 Madison Avenue, New York, NY 10017, by telephone at (212) 518-5548, or by email at GSEquityProspectusDelivery@guggenheimpartners.com.
This press release shall not constitute an offer to sell or the solicitation of an offer to buy, nor shall there be any sale of these securities in any state, province, territory or jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state, province, territory or jurisdiction.
About Kymera
Kymera Therapeutics is a biopharmaceutical company focused on advancing the field of targeted protein degradation by accessing the body’s innate protein recycling machinery to degrade dysregulated, disease-causing proteins to address previously intractable disease targets.
Forward-Looking Statements
This press release includes certain disclosures that contain “forward-looking statements,” including, without limitation, statements regarding Kymera’s expectations regarding the commencement of trading of its shares on the Nasdaq Global Market, the completion and timing of the closing of the offering and the anticipated gross proceeds from the offering. Forward-looking statements are based on Kymera’s current expectations and are subject to inherent uncertainties, risks and assumptions that are difficult to predict. Factors that could cause actual results to differ include, but are not limited to, risks and uncertainties related to the satisfaction of customary closing conditions and the completion of the offering, and the risks inherent in biopharmaceutical product development and clinical trials. These and other risks and uncertainties are described more fully in the section titled “Risk Factors” in the final prospectus related to the offering to be filed with the Securities and Exchange Commission. Forward-looking statements contained in this announcement are made as of this date, and Kymera undertakes no duty to update such information except as required under applicable law.
Contact:
Investors@kymeratx.com
Bruce Jacobs
Chief Financial Officer
(857) 285-5314

Integrates new capabilities for pharmaceutical manufacturers, PBMs and payers to simplify rebate negotiation and payment management along with real-time insights into contract performance.
PHILADELPHIA, July 20, 2020 /PRNewswire/ -- HealthVerity, the leader in privacy-protected data exchange, today announced that it has acquired Curisium, an innovator in cloud-based drug rebate processing and contract performance management. The combination of HealthVerity and Curisium will deliver a more complete and responsive infrastructure for high-governance, privacy- compliant data sharing between pharmaceutical manufacturers, pharmacy beneits managers (PBMs) and payers.
Current Curisium clients leverage cutting-edge technologies that streamline and consolidate contract negotiation, rebate validation and invoicing through a fully managed SaaS solution. Built upon a highly responsive, rules-driven solution, Curisium's platform is the operating system that delivers automated workflows standardizing contracting, adjudication, and payment processes, and replacing disparate analog processes and spreadsheets. HealthVerity is now positioned to deliver technologies that can dramatically improve the eficiency and transparency of the cost and complications of patients on a drug. For pharmaceutical manufacturers, brands can more eficiently track the funds flow on existing contracts, better incorporate third party data to understand the healthcare journey of patients on therapy and leverage more advanced technologies to optimize the design and ultimate success of value-based contracts. For PBMs and payers, HealthVerity now represents a turnkey solution for simpliication of the contracting and adjudication process, enabling privacy-protected data exchange that establishes trust between counterparties in pursuit of cost-effective treatment pathways, advanced analytics and superior patient outcomes.
HealthVerity has emerged as a leader and key partner to nearly 75% of top pharmaceutical manufacturers and payers, having fundamentally changed the way that key healthcare organizations secure, exchange and synthesize HIPAA-protected information at scale. With software and APIs that enable access to de-identiied data for more than 330 million individuals across the largest healthcare and consumer data ecosystem in the US, HealthVerity is well positioned to enable clients to signiicantly improve the way counterparties can derive more actionable clinical and inancial impacts.
"Curisium had the vision to build an innovative framework and SaaS platform that allows parties to seamlessly negotiate and visualize contractual relationships around drug utilization, which is fundamentally similar to the HealthVerity approach to making our data ecosystem available for data sharing or analytic applications," said Andrew Kress, CEO of HealthVerity. "The idea of creating a frictionless, standardized, scalable architecture to support privacy-protected data workflows is complementary to everything that we do. In light of the disruptions due to COVID-19 as well as the potential of proposed changes by CMS to value-based agreements, we believe this is an ideal time to scale additional tools that provide our data partners and our healthcare clients an entirely new way to transact around speciic contracts."
"We are excited to join HealthVerity and to continue our mission of empowering life sciences companies and payers with a more agile, progressive approach to healthcare contracting," said Peter Kim, Co-founder and CEO of Curisium. "Our clients will now have access to clinically relevant outcomes that have beneitted from these healthcare contracts, in addition to a greater understanding of the impact on patient lives. We see HealthVerity as being well positioned to become the premier network for privacy-protected data exchange across biopharma, payers, PBMs and pharmacies and we're excited to join forces in this cause."
Financial terms of the transaction were not released. HealthVerity was represented by Blank Rome LLP while Curisium was represented by Cooley LLP.
About HealthVerity
Powering the largest US healthcare and consumer data ecosystem, combined with best-in-class management and privacy solutions, HealthVerity is helping answer healthcare's most critical questions. Our technology platform serves as the foundation for the rapid creation, exchange and management of healthcare and consumer data in a fully-interoperable, privacy-protecting manner. Advantaged by highly sophisticated identity resolution and matching capabilities, HealthVerity is on a mission to increase transparency, forge interoperability and activate deeper insights. To learn more about HealthVerity's technology platform, visit www.healthverity.com.
Contact:
Abigail Stockwell
Senior Director, Marketing
HealthVerity
astockwell@healthverity.com
SOURCE HealthVerity
Related Links
- Kymera to receive $150 million upfront with more than $2 billion in potential milestones plus royalty payments
- Kymera to retain option during clinical development to participate equally in US cost and profit sharing
CAMBRIDGE, Mass. (July 9, 2020) – Kymera Therapeutics Inc. today announced the company has entered into a multi-program strategic collaboration with Sanofi (NASDQ: SNY) to develop and commercialize first-in-class protein degrader therapies targeting IRAK4 in patients with immune-inflammatory diseases. The companies will also partner on a second earlier stage program. Kymera will receive $150 million in cash upfront and may receive more than $2 billion in potential development, regulatory and sales milestones, as well as significant royalty payments. Kymera retains the option to participate in US development and commercialization for both programs. This includes the ability to participate equally in the costs, profits and losses after opt-in, and to co-promote partnered products in the US.
“This is an important collaboration for both companies and for the field of targeted protein degradation,” said Nello Mainolfi, Ph.D., co-founder, President and CEO of Kymera Therapeutics. “Kymera is becoming a fully integrated biotechnology company advancing a pipeline of novel therapies with the potential to transform treatment paradigms. We are excited to partner with Sanofi, an organization with world-class drug development and commercialization capabilities, to ensure maximal patient impact from two of our programs across multiple disease indications, while enabling Kymera to invest in key strategic areas to realize the broad potential of protein degrader therapies.”
Under terms of the collaboration, Sanofi will make an upfront payment of $150 million in cash to Kymera for global rights to develop its small molecule IRAK4 protein degraders in inflammation and immunology indications, and a second earlier stage undisclosed program. IRAK4 is believed to play a key role in multiple immune-inflammatory diseases, including hidradenitis suppurativa, atopic dermatitis and rheumatoid arthritis. Kymera will advance the IRAK4 program through Phase 1 clinical trials; Sanofi will assume clinical development and commercialization responsibilities thereafter. Sanofi will lead all clinical development activities for the second program. Kymera will have the option to participate in the development of both programs in the US during clinical development. Kymera will retain global rights to its IRAK4 program in oncology indications.
IRAK4 is a key protein involved in inflammation mediated by the activation of toll-like receptors (TLRs) and IL-1 receptors (IL-1Rs). While TLR and IL-1R signaling via IRAK4 is involved in the normal immune response, aberrant activation of these pathways is the underlying cause of multiple immune-inflammatory conditions. In pre-clinical studies, Kymera has shown oral daily administration of an IRAK4 degrader can lead to complete knockdown of IRAK4 in skin and immune cells in higher species and is well tolerated. Data presented at the most recent annual meetings of the American College of Rheumatology and the European Hidradenitis Suppurativa Foundation showed potent anti- inflammatory activity in both in vitro and in vivo preclinical models."
“Targeted protein degrada on is an exci ng modality. Kymera has developed an incredible drug discovery engine producing protein degraders with compelling and dieren ated pharmacology against targets that, to date, have not been op mally addressed with other therapeu c modali es,” said John Reed, Global Head of Research & Development at Sano. “We are excited to partner with the Kymera team to advance a new genera on of rst-in-class therapies with the poten al to eliminate underlying drivers of disease.”
Aquilo Partners, L.P. acted as Financial advisor to Kymera on this transac on.
# # #
About Kymera Therapeutics
Kymera Therapeutics is a biotechnology company pioneering a transformative new approach to treating previously untreatable diseases. The company is advancing the field of targeted protein degradation, accessing the body’s innate protein recycling machinery to degrade dysregulated, disease-causing proteins. Kymera’s Pegasus targeted protein degradation platform harnesses the body’s natural protein recycling machinery to degrade disease-causing proteins, with a focus on un-drugged nodes in validated pathways currently inaccessible with conventional therapeutics.
Kymera is accelerating drug discovery with an unmatched ability to target and degrade the most intractable of proteins, and advance new treatment options for patients. For more information, visit www.kymeratx.com.
About Sanofi
Sanofi is dedicated to supporting people through their health challenges. We are a global pharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions. With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe. Sanofi, empowering life. For more information, visit www.sanofi.com.
Sam Marwaha joins as Chief Commercial Officer; Company to deploy virtual
health programs for its network of nearly 4 million individuals later this year
SAN MATEO, Calif., July 1, 2020 — Evidation Health today announced the close of $45 million in Series D funding to power the expansion of its privacy-first, direct-to-person research platform, Achievement, to include virtual health. The company will use this new capital to build upon its existing evidence generation capabilities to offer real-time insights and digital tools to its network of nearly 4 million individuals.
B Capital Group led the round, which includes new investors McKesson Ventures and Section 32 as well as existing investors Revelation Partners, Rethink Impact, and SV Health Investors. B Capital’s Adam Seabrook has joined Evidation’s Board of Directors. Sam Marwaha, Senior Partner at Boston Consulting Group and long-time advisor to Evidation, joins the company as Chief Commercial Officer.
The company will leverage this investment to continue to rapidly grow its study solutions business and offer programs that enable Achievement members to gain clinically actionable insights into their health. These include relevant digital interventions, treatments, and tools to help individuals understand and take advantage of the latest evidence, in partnership with their care teams. The first of these new virtual health offerings is slated for release later this year. Evidation will continue to expand and diversify the Achievement population — already the largest and most demographically and geographically diverse connected cohort in the United States — to serve additional at-risk and disease-specific cohorts. Achievement already includes participants residing in all 50 states and in 9 out of every 10 ZIP codes nationwide.
“Participation in research is an activator for individuals to better understand their health and seek care proactively,” said Evidation CEO Deborah Kilpatrick, Ph.D. “The natural next step for Evidation is to decrease the latency between evidence and action by offering virtual health programs, while still keeping privacy and consent at the forefront. We are especially excited to have Sam Marwaha, a pioneer in real-world evidence and an experienced digital health strategist and operator, join us to lead our commercial endeavors as the company ushers in a new era of self-activated care.”
Marwaha will lead Evidation’s commercial strategy as it brings its new offerings to market. A 20-year industry veteran, Sam will strengthen and broaden partnerships with life sciences companies as they innovate with patients, providers, and payers. He will also scale Evidation’s marketing and business development teams and shape new opportunities to help people achieve better outcomes through virtual health.
“Evidation is a learning engine directly connected to four million people, making it possible to understand real-life indicators of health quality and disease progression, reflecting behavioral and social factors,” said Marwaha. “Evidation can drive personalized, consented engagement with individuals in the safety and convenience of their homes, empowering them to be partners in their own health.”
“Healthcare needs Evidation more than ever before,” said Raj Ganguly, co-Founder and Managing Partner at B Capital Group. “Evidation’s technology has the potential to realize new ways for individuals, providers, biopharma, and payers to understand and react to health in real life. Today’s announcement marks the company’s next step toward achieving that goal and we are pleased to continue our partnership with the company as investors.”
“We see tremendous value in generating real-world data for both clinical development and commercial life sciences use cases, ensuring the best possible understanding of a population inside and outside the clinic,” said Carrie Hurwitz Williams of McKesson Ventures. “The discipline, rigor and scale with which Evidation has built their technology makes it possible to garner relevant real-world data points and transform them into meaningful insights for pharma.”
This funding and C-suite hire caps off a fast-paced growth period for Evidation. Eight of the top 10 global biopharma companies rely on Evidation for person-generated health data (PGHD), including passive sensor data, electronic patient reported outcomes (ePROs), and at-home diagnostics. Johnson & Johnson, Apple, and Evidation recently launched the ongoing Heartline Study, the largest randomized trial in the history of cardiovascular disease. Evidation is also working with a diverse cross section of partners in industry, academia, government, and civil society, including the Icahn School of Medicine at Mount Sinai, the Biomedical Advanced Research and Development Authority (BARDA), and the Bill & Melinda Gates Foundation, on efforts to better detect, characterize and predict COVID-19 using PGHD.
“COVID-19 has shown us that accelerating real-world evidence development is critical,” said Mike Pellini, MD, Managing Partner of Section 32 and former CEO and Chairman of Foundation Medicine. “Evidation has led the way in health measurement and has proven out the potential of person-generated health data. The company is optimally positioned to lead the way into a new era of individualized healthcare.”
About Evidation
Evidation measures health in everyday life and enables anyone to participate in ground-breaking research and health programs. Built upon a foundation of user privacy and control over permissioned health data, Evidation’s Achievement platform is trusted by millions of
individuals—generating data with unprecedented speed, scale, and rigor. We partner with leading healthcare companies to understand health and disease outside the clinic walls. Guided by our mission to enable and empower everyone to participate in better health outcomes, Evidation is working to bring people individualized, proactive, and accessible healthcare—faster. Founded in 2012, Evidation Health is headquartered in California with additional offices around the globe. To learn more, visit evidation.com, or follow us on Twitter @evidation.
Media Contact
press@evidation.com
CAMBRIDGE, Mass., June 10, 2020 (GLOBE NEWSWIRE) -- Fulcrum Therapeutics, Inc. (Nasdaq: FULC) today announced the closing of a $68.5 million private placement pursuant to a securities purchase agreement with a group of institutional investors and accredited investors.
The private placement includes a mix of new and existing investors, including EcoR1 Capital, LLC, Alyeska Investment Group, L.P., Boxer Capital, Casdin Capital LLC, Perceptive Advisors LLC, Samsara BioCapital, Monashee Investment Management LLC and Foresite Capital.
SVB Leerink acted as the exclusive placement agent to the Company in connection with the private placement.
In the private placement, the Company sold 4,029,411 shares of common stock at a price of $17.00 per share. The private placement priced on June 8, 2020.
The Company expects to use net proceeds from the private placement to fund research and development expenses, including the ongoing clinical development of losmapimod for facioscapulohumeral muscular dystrophy (FSHD), the initiation of clinical development of losmapimod for COVID-19 pending review of the Company’s recently filed investigational new drug application by the U.S. Food and Drug Administration, the advancement of its hemoglobinopathies program into clinical development and the advancement of both its discovery efforts and product engine capabilities, as well as working capital and other general corporate purposes.
The securities sold in the private placement have not been registered under the Securities Act of 1933, as amended (the “Securities Act”), or any state or other applicable jurisdiction’s securities laws, and may not be offered or sold in the United States absent registration or an applicable exemption from the registration requirements of the Securities Act and applicable state or other jurisdictions’ securities laws. The Company has agreed to file a registration statement with the U.S. Securities and Exchange Commission (the “SEC”) registering the resale of the shares of common stock issued in the private placement no later than the 15th day after the date of the securities purchase agreement for the private placement.
This press release shall not constitute an offer to sell or the solicitation of an offer to buy these securities, nor shall there be any offer, solicitation or sale of these securities in any jurisdiction in which such offer, solicitation or sale would be unlawful. Any offering of the securities under the resale registration statement will only be made by means of a prospectus.
About Fulcrum Therapeutics
Fulcrum Therapeutics is a clinical-stage biopharmaceutical company focused on improving the lives of patients with genetically defined rare diseases in areas of high unmet medical need. Fulcrum’s proprietary product engine identifies drug targets which can modulate gene expression to treat the known root cause of gene mis-expression. The company has advanced losmapimod to Phase 2 clinical development for the treatment of facioscapulohumeral muscular dystrophy (FSHD) and has completed extensive pre-clinical research for FTX-6058 for the treatment of sickle cell disease and beta-thalassemia.
Cautionary Note Regarding Forward-Looking Statements
This press release contains forward-looking statements within the meaning of The Private Securities Litigation Reform Act of 1995. Such forwardlooking statements include, but are not limited to, those regarding: the anticipated use of proceeds from the private placement; the filing of a registration statement to register the resale of the shares issued and sold in the private placement; and the Company’s plans, strategies and prospects for its business, including the Company’s plans to evaluate losmapimod as a potential treatment for COVID-19. All statements, other than statements of historical facts, contained in this press release, including statements regarding the Company’s strategy, future operations, future financial position, prospects, plans and objectives of management, are forward-looking statements. The words “anticipate,” “believe,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “potential,” “predict,” “project,” “should,” “target,” “will,” “would” and similar expressions are intended to identify forward-looking statements, although not all forward-looking statements contain these identifying words. Any forward-looking statements are based on management’s current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in, or implied by, such forward-looking statements. These risks and uncertainties include, but are not limited to, risks associated with Fulcrum’s ability to obtain and maintain necessary approvals from the FDA and other regulatory authorities; continue to advance its product candidates in clinical trials; replicate in later clinical trials positive results found in preclinical studies and early-stage clinical trials of losmapimod and its other product candidates; advance the development of its product candidates under the timelines it anticipates in current and future clinical trials, if at all; obtain, maintain or protect intellectual property rights related to its product candidates; manage expenses; and raise the substantial additional capital needed to achieve its business objectives. For a discussion of other risks and uncertainties, and other important factors, any of which could cause the Company’s actual results to differ from those contained in the forward-looking statements, see the “Risk Factors” section, as well as discussions of potential risks, uncertainties and other important factors, in the Company’s most recent filings with the Securities and Exchange Commission. In addition, the forward-looking statements included in this press release represent the Company’s views as of the date hereof and should not be relied upon as representing the Company’s views as of any date subsequent to the date hereof. The Company anticipates that subsequent events and developments will cause the Company’s views to change. However, while the Company may elect to update these forwardlooking statements at some point in the future, the Company specifically disclaims any obligation to do so.
Contact:
Investors:
Christi Waarich
Director, Investor Relations and
Corporate Communications
617-651-8664
cwaarich@fulcrumtx.com
Stephanie Ascher
Stern Investor Relations, Inc.
stephanie.ascher@sternir.com
212-362-1200
Media:
Kaitlin Gallagher
Berry & Company Public Relations
kgallagher@berrypr.com
212-253-8881
Collaboration aims to develop predictive algorithm for symptoms of COVID-19 by monitoring first responders
SAN MATEO, Calif., June 4, 2020 – Evidation Health today announced a new effort to develop an early warning algorithm to detect symptoms of COVID-19 and to understand susceptibility to infection, funded by the Biomedical Advanced Research and Development
Authority (BARDA), part of the Office of the Assistant Secretary for Preparedness and Response at the U.S. Department of Health and Human Services (HHS) and the Bill & Melinda Gates Foundation.
The Evidation platform will analyze behavior, including sleep and activity patterns, alongside self-reported symptoms for 300 people at high risk of developing COVID-19. This builds on Evidation’s current, ongoing 150,000-person nationwide initiative tracking people’s health and attitudes during the pandemic, COVID-19 Pulse.
“Many infected individuals are asymptomatic but still able to spread the virus, making efforts to prevent and slow transmission of COVID-19 difficult,” said Luca Foschini, Ph.D., Evidation’s co-founder and chief data scientist. “This initiative will use novel behavioral and physiological data to more effectively identify when and where people may contract COVID-19 and can potentially enable real-time interventions to limit spread and monitor outcomes.”
The analysis, performed in collaboration with non-profit 4YouandMe, will use de-identified data generated by self-reporting and wearable devices to track symptoms of COVID-19 in those at particularly high risk, including health care workers and other first responders, in order to better understand susceptibility to SARS-CoV-2 infection. One potential outcome of this work is an early warning algorithm to help individuals better understand and monitor their respiratory disease symptoms and take precautions against their spread.
“The ability to self-monitor and be informed of health status will empower Americans in their decisions to help slow the spread of this pandemic and improve health outcomes for people with COVID-19,” said BARDA Acting Director, Gary Disbrow, Ph.D. “This pilot study is not only an early step in demonstrating the utility of models developed using person-generated health data but also may provide data to better understand the varied symptoms of COVID-19.”
This program follows Evidation’s work with BARDA to monitor individuals for respiratory infections, such as influenza. Evidation’s existing research on influenza utilizes person-generated health data and population-based models with the goal of improving real-time respiratory infection monitoring at the individual and population level. BARDA is contributing a $720,000 award as part of BARDA’s COVID-19 Rapidly Deployable Capabilities program to identify and pilot near-term innovative solutions for COVID-19. Support from the Bill & Melinda Gates Foundation is from the $250 million the foundation has committed to address the COVID-19 pandemic.
About Evidation
Evidation measures health in everyday life and enables anyone to participate in ground-breaking research and health programs. Built upon a foundation of user privacy and control over permissioned health data, Evidation's Achievement platform is trusted by millions of
individuals—generating data with unprecedented speed, scale, and rigor. We partner with leading healthcare companies to understand health and disease outside the clinic walls. Guided by our mission to enable and empower everyone to participate in better health outcomes, Evidation is working to bring people individualized, proactive, and accessible healthcare—faster. Founded in 2012, Evidation Health is headquartered in California with additional offices around the globe. To learn more, visit evidation.com, or follow us on Twitter @evidation.
About HHS, ASPR, and BARDA
HHS works to enhance and protect the health and well-being of all Americans, providing for effective health and human services and fostering advances in medicine, public health, and social services. The mission of ASPR is to save lives and protect Americans from 21st century health security threats. Within ASPR, BARDA invests in the advanced research and development, acquisition, and manufacturing of medical countermeasures – vaccines, drugs, therapeutics, diagnostic tools, and non-pharmaceutical products needed to combat health security threats. To date, 54 BARDA-supported products have achieved regulatory approval, licensure or clearance. Learn more about preparing for and responding to public health emergencies, from new infectious diseases to natural disasters and bioterrorism, by visiting the HHS public health emergency website, www.phe.gov. For more information on partnering with BARDA on developing medical countermeasures, visit www.medicalcountermeasures.gov.
Media Contact
press@evidation.com

- Proprietary tissue-targeted complement platform and best-in-class IL-7R antibody technology to treat patients with severe autoimmune and inflammatory disease; clinical trials begin in 2020
- Previously closed $46M Series A led by Atlas Venture included OrbiMed Advisors, Abingworth, and Sanofi Ventures
- Company appoints Michael Broxson as CEO; co-founder Shelia Violette as Chief Scientific Officer and President of Research
Cambridge, Mass. (May 27, 2020) – Q32 Bio, a biotechnology company developing biologics to restore healthy immune regulation, today announced the company’s pipeline and plans to enter the clinic this year with its lead candidate. Enabled by a $46M Series A financing led by Atlas Venture, and a world class team and Scientific Advisory Board, Q32 Bio is advancing a portfolio of biologics targeting the body’s innate and adaptive immune systems. The company has a robust product pipeline including a monoclonal antibody antagonist of the interleukin-7 receptor (IL-7R), and a complement therapeutics platform that has generated fusion proteins that downregulate complement activity specifically in disease-affected tissues. The company is led by industry veterans Michael Broxson as Chief Executive Officer, and co-founder, Shelia Violette, Ph.D., as Chief Scientific Officer and President of Research.
Q32 Bio was seeded and incubated by Atlas Venture with foundational science from renowned researchers in immunology Michael Holers, M.D. and Joshua Thurman, M.D., both from the University of Colorado, and Steven Tomlinson, Ph.D. from the Medical University of South Carolina. Dr. Holers serves as chairman of the Scientific Advisory Board, which is comprised of distinguished global experts in autoimmune and inflammatory disease.
The subsequent Series A financing included Atlas Venture, OrbiMed Advisors, Abingworth, Sanofi Ventures, University of Colorado and Children’s Hospital Colorado Center for Innovation.
“Autoimmune and inflammatory diseases are driven by dysregulation of the immune response,” said Mr. Broxson. “Q32 Bio has a preeminent team with expertise in both innate and adaptive immunity, a board of directors and scientific advisory board made up of leaders in our field, and a syndicate of blue-chip investors. This gives us a running start in developing therapies that may improve and save lives. I am thrilled to join Q32 Bio and look forward to advancing our first two immune-regulating therapies into first-in-human studies planned for 2020 and 2021.”
Integrated Approach to Restoring Healthy Immune Regulation
Q32 Bio’s most advanced program, ADX-914, is a fully human anti-IL-7R antibody licensed from Bristol Myers Squibb (NYSE: BMY) that re-regulates adaptive immune function. The cytokine IL-7 has demonstrated the ability to drive several T-cell mediated pathological processes. It stimulates T-effector and T-memory cells, lowering the threshold-response to disease related antigens; inhibits the immunosuppressive function of T-regulatory cells; and promotes pathogenic autoantibody production. Inhibition of IL-7R signaling has the potential to durably and safely restore healthy immune regulation in numerous autoimmune and inflammatory diseases. Q32 Bio is advancing plans for the first-in-human trial of ADX-914 while continuing to monitor the potential impact of COVID-19 on clinical operations. Pending confirmation that trial sites are able to operate and enroll patients safely, a biomarker-guided ADX-914 Phase 1 trial is planned to start in late 2020.
Q32 Bio is developing ADX-914 (formerly known as BMS-986265) as part of an exclusive worldwide licensing agreement with Bristol Myers Squibb. Under the terms of the agreement, Bristol Myers Squibb received an upfront payment and became a minority shareholder of Q32 Bio. Bristol Myers Squibb has the potential to earn additional milestone payments subject to the achievement of certain development, regulatory and sales-based milestones, as well as tiered annual net sales royalties. Q32 Bio is solely responsible for all future development and commercialization costs of ADX-914.
Q32 Bio’s lead program in innate immunity, ADX-097, is based on a groundbreaking platform providing tissue-targeted regulation of the complement system. Complement is an integral component of the innate immune system that provides a first line of defense for clearing pathogens and removing damaged cells. In a wide range of autoimmune and inflammatory diseases the complement system becomes hyperactivated, causing the immune system to attack and damage otherwise healthy tissue. Q32 Bio’s proprietary and unique platform has yielded a pipeline of novel protein therapeutics that provide potent inhibition of complement in diseased tissues without long-term systemic blockade – a key differentiator versus current complement therapeutics. Q32 Bio has initiated IND-enabling activities for ADX-097 and first-in-human dosing is planned for 2H 2021.
“Given their broad roles in mediating immunity, the complement system and IL-7 signaling pathways are high potential arenas for drug development,” said Dave Grayzel, MD, Partner, Atlas Venture and chairman of the Q32 Bio Board of Directors. “With a best-in-class IL-7R antibody and fusion proteins designed to target the complement system at sites of activation in a unique and differentiated manner, Q32 Bio is well positioned to make a meaningful impact for patients with serious inflammatory and autoimmune diseases.”
Experienced Leadership Team
Michael Broxson brings over 20 years of industry experience to Q32 Bio. He joined from Goldfinch Bio, where he was Chief Business and Operating officer. Prior to Goldfinch he served in leadership roles in business development, new product planning, strategy and
finance at Takeda Pharmaceuticals. He holds BA and MSPH degrees in Toxicology from Tulane University and an MBA from the University of Chicago’s Booth School of Business and is a CFA® charterholder.
Shelia Violette, Ph.D., joined Q32 Bio from Atlas Venture, where she was Entrepreneur in Residence and initially co-led the formation of Q32 Bio. Previously she served in leadership roles in the Immunology Research group at Biogen, joining Stromedix as Vice President of Research and returning to Biogen as Vice President of Research, spearheading the tissue injury and fibrosis therapeutic area. She holds a Ph.D. in Pharmacology from Yale University.
About Q32 Bio
Q32 Bio is a biotechnology company developing therapies targeting powerful regulators of the innate and adaptive immune systems, to re-balance immunity in severe autoimmune and inflammatory diseases. Q32 Bio’s lead programs, focused on the IL-7R pathway and complement system, address immune dysregulation to help patients take back control of their lives. For more information, please visit www.Q32bio.com.
About Atlas Venture
Atlas Venture is a leading biotech venture capital firm. With the goal of doing well by doing good, the company has been building breakthrough biotech startups for over 25 years. Atlas works side by side with exceptional scientists and entrepreneurs to translate high impact science into medicines for patients. Our seed-led venture creation strategy rigorously selects and focuses investment on the most compelling opportunities to build scalable businesses and realize value. For more information, please visit www.atlasventure.com.
About Abingworth
Abingworth is a leading transatlantic life sciences investment firm. We help transform cutting-edge science into novel medicines by providing capital and expertise to top caliber management teams and building world-class companies. With offices in Menlo Park (California), Boston and London, Abingworth has invested in approximately 160 life science companies, leading to more than 40 M&A/exits and over 65 IPOs since 1973.
About OrbiMed
OrbiMed is a leading healthcare investment firm, with $13 billion in assets under management. OrbiMed invests globally across the healthcare industry, from start-ups to large multinational corporations, utilizing a range of private equity funds, public equity funds, and royalty/credit funds. OrbiMed maintains offices in New York City, San Francisco, Shanghai, Hong Kong, Mumbai and Herzliya. OrbiMed seeks to be a capital provider of choice, providing tailored financing solutions and extensive global team resources to help build world-class healthcare companies.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi. Sanofi Ventures invests in early-stage biotech and digital health companies with innovative ideas and transformative new products and technologies of strategic interest to Sanofi. Among these areas are oncology, immunology, rare diseases, vaccines, potential cures in other core areas of Sanofi’s business footprint, and digital health solutions. For more information, visit www.sanofiventures.com.
Media Contact
Arleen Goldenberg
Verge Scientific Communications
agoldenberg@vergescientific.com
917.548.1582
Financing will support i20 Therapeutics’ growth and R&D of platform for oral delivery of injectable biologic drugs
BOSTON, MA—April 9, 2020—i2O Therapeutics, an innovative biotech company developing a platform for oral delivery of traditionally injectable biological drugs, announced today it has raised a $4 million seed funding round led by Sanofi Ventures and JDRF T1D Fund.
Founded by a team of researchers from Harvard University, i2O Therapeutics is focused on the development of safe and effective oral formulations of therapies that are conventionally limited to injections, e.g. biologics, large molecules, and peptide-based pharmaceuticals such as insulin. The foundational technology has been exclusively licensed to i2O by Harvard’s Office of Technology
Development. The company’s innovative platform enables drugs that traditionally wouldn’t survive the hostile environment of the digestive system to pass through safely by utilizing a unique coating that dissolves in the small intestine, thereby releasing the active drug. I2O’s initial focus is on developing a novel oral formulation for GLP-1 analogs.
The technology was initially developed in the Harvard lab of Samir Mitragotri, PhD, who is Hiller Professor of Bioengineering and Hansjorg Wyss Professor of Biologically Inspired Engineering at Harvard John A. Paulson School of Engineering and Applied Sciences and a Core Faculty member at Harvard’s Wyss Institute for Biologically Inspired Engineering.
“Our technology has the potential to enable the oral delivery of high-value drugs in a safer, more effective and patient-friendly way and also by easing the treatment burden for dozens of therapeutics that were previously restricted to intravenous or subcutaneous delivery,” commented Mitragotri, Cofounder of i2O Therapeutics.
“The support and partnership of Sanofi Ventures and JDRF T1D Fund marks a major milestone for i2O, and potentially millions of people around the world. This round of financing will enable us to rapidly expand our team and ramp up research and development at i2O as we seek to create the next generation of oral peptide and protein-based therapies,” commented Ravi Srinivasan, PhD, Co-founder of i2O Therapeutics.
“i2O Therapeutics is developing an extremely promising new platform for oral biologics with the potential to significantly ease treatment burden for countless patients who rely on drugs that can only be administered via injection,” said Christopher Gagliardi, PhD, Director of Investments at Sanofi Ventures.
“We are excited to partner with i2O Therapeutics, whose platform has the potential to revolutionize the way people with diabetes manage their disease,” commented Katie Ellias, Managing Director of the JDRF T1D Fund. “The possibility of an oral insulin product, among other exciting applications of the i2O platform, represents a significant commercial opportunity and more importantly, has the potential to improve glycemic management and decrease hypoglycemia risk over today’s injectable insulins.”
Both Christopher Gagliardi and Katie Ellias will join the i2O Board of Directors.
About i2O Therapeutics
i2O Therapeutics is a biotechnology company developing safe and effective oral formulations of therapies traditionally limited to injections. Using an innovative ionic liquid technology, this platform leverages the benefits of protecting the drug cargo while also transiently enhancing permeation across the epithelial lining when administered orally. i2O is focused on creating the next generation of oral peptide and protein-based therapies. Visit us at www.i2OBio.com.
About JDRF T1D Fund
The JDRF T1D Fund (https://t1dfund.org/) is a venture philanthropy fund accelerating life-changing solutions to cure, prevent and treat type 1 diabetes (T1D) through catalytic equity investments. Through its investments in partnership with private capital, including venture capital, corporations and foundations, the T1D Fund seeks to attract the private investment necessary to advance therapeutics, devices, diagnostics, and vaccines into the hands of those living with T1D. The T1D Fund invests in areas strategically aligned with JDRF, the leading global organization funding T1D research, with an exclusive focus on supporting the best commercial opportunities. The T1D Fund reinvests any realized gains into new investments to further its mission.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi. Sanofi Ventures invests in early-stage biotech and digital health companies with innovative ideas and transformative new products and technologies of strategic interest to Sanofi. Among these areas are oncology, immunology, rare diseases, vaccines, potential cures in other core areas of Sanofi’s business footprint, and digital health solutions. For more information, visit www.sanofiventures.com.
Contact
Gregory Johnson, PhD
MacDougall
Gjohnson@macbiocom.com
- First patient dosed in Phase 1 clinical trial evaluating IL-7R antibody ADX-914
- Financing round co-led by OrbiMed Advisors and Acorn Bioventures fuels advancement of portfolio
Cambridge, Mass. (October 29, 2020) – Q32 Bio, a biotechnology company developing biologic therapeutics to restore healthy immune regulation, today announced the completion of a $60 million Series B financing co-led by OrbiMed Advisors and Acorn Bioventures. The company also announced the commencement of a Phase 1 clinical trial evaluating its best-in-class IL-7R antibody ADX-914, a significant milestone marking Q32 Bio’s entry into clinical-stage development.
Proceeds from the Series B financing will support the advancement of ADX-914 through demonstration of proof-of-mechanism and propel the company’s groundbreaking tissue-targeted complement regulation platform. ADX-097 is the first fusion protein generated by Q32’s complement platform and is expected to enter Phase 1 in the fourth quarter of 2021. The financing also enables further refinement and expansion of both the company’s platform and pipeline.
“The closing of our Series B financing and entry into the clinic are pivotal milestones for Q32 Bio,” said Michael Broxson, CEO of Q32 Bio. “The initiation of the Phase 1 trial for ADX-914 represents an important achievement and a significant step toward bringing a powerful new treatment option to patients with autoimmune disease. We expect to continue to make major advances over the coming year as we move ADX-097 into the clinic.”
In addition to Orbimed Advisors and Acorn Bioventures, Series B investors include Osage University Partners, Atlas Venture, Abingworth, Sanofi Ventures, University of Colorado and Children’s Hospital Colorado Center for Innovation. In conjunction with the Series B financing, Isaac Manke (Acorn Bioventures) and Diyong Xu (OrbiMed Advisors) will join the Q32 Bio Board of Directors.
“We are thrilled to partner with a stellar group of investors to support Q32’s novel therapeutic platform,” said Stephen Squinto, Ph.D., Executive Partner, OrbiMed Advisors, member of the Q32 Board of Directors, and Scientific Advisor to the company. “With a premier investor syndicate and world class leadership team, Q32 Bio is well positioned to advance a differentiated portfolio of therapies targeting important regulators of both adaptive and innate immunity.”
“Q32 Bio is an ideal investment for Acorn Bioventures, which provides committed capital through multiple financing rounds to innovative and entrepreneurial biotech companies,” said Isaac Manke, Acorn Bioventures partner and Q32 Bio Board member. “We are delighted to partner with the company’s outstanding leadership team in Q32’s mission to create the next generation of immune-modulating therapies for patients.”
About Q32 Bio
Q32 Bio is a clinical stage biotechnology company developing therapies targeting powerful regulators of the innate and adaptive immune systems to re-balance immunity in severe autoimmune and inflammatory diseases. Q32 Bio’s lead programs, focused on the IL-7R pathway and complement system, address immune dysregulation to help patients take back control of their lives.
Q32 Bio’s approach centers on restoring healthy immune regulation in autoimmune and inflammatory disease, with a focus on pathways that are critical modulators of innate and adaptive immunity. The first-in-human trial for the company’s most advanced program, ADX-914, a fully human anti-IL-7R antibody, is designed to evaluate safety and tolerability and demonstrate proof-of-mechanism. IL-7 has been genetically and biologically validated to drive several T cell-mediated pathological processes in numerous autoimmune diseases.
Q32 Bio’s lead program for innate immunity, ADX-097, is based on a pioneering approach enabling tissue-targeted regulation of the complement system without long-term systemic blockade – a key differentiator versus current complement therapeutics. First-in-human dosing of ADX-097 is planned for 2H 2021.
For more information, please visit www.Q32bio.com.
###
Media Contact
Arleen Goldenberg
Verge Scientific Communications
agoldenberg@vergescientific.com
917.548.1582
PARIS and NEW YORK – November 20, 2019 - Sanofi announced today an enterprise- wide collaboration with health care technology company Aetion that will integrate Sanofi’s real-world data platform, DARWIN, with the Aetion Evidence Platform® with the objective of advancing more efficient use of real-world evidence (RWE), facilitating regulatory-grade studies with deep transparency, and unlocking access to new real-world data.
Both companies have invested in RWE platforms, recognizing the pressing need for accurate, fast, and cost-effective research and the important role RWE could play in meeting this need. Sanofi’s DARWIN compiles and analyzes de-identified data from hundreds of millions of patients across disease states, while Aetion’s platform analyzes real-world data to produce transparent, rapid, and scientifically validated answers about the effectiveness, safety, and value of drugs. By combining these platforms, Sanofi is seeking to elevate its capabilities in conducting regulatory-grade analytics, opening new doors for the development and application of medical treatments.
“Today marks another important step in Sanofi’s digital transformation,” said Bernard Hamelin, MD, MSc, MBA, Global Head of Medical Evidence Generation, Sanofi. “By integrating these platforms, we strive to make faster, more informed decisions with the potential to lead to first-in-class and best-in-class treatments that could change the practice of medicine.”
Real-world evidence offers a view of clinical practice outside of the experimental setting, providing an opportunity to inform clinical trial development and supplement trial data with evidence of actual product use in the health care system.
“Our work with Sanofi further validates the value and potential for real-world evidence in drug development,” said Carolyn Magill, Chief Executive Officer of Aetion. “Our companies share a common goal of using the best available data to get the right treatment to the right patient as quickly and efficiently as possible.”
This collaboration between Sanofi and Aetion demonstrates leadership during a critical time. Real-world evidence is expected to play a key role in transforming the health care ecosystem, with the U.S. Food and Drug Administration (FDA) recently prioritizing efforts to incorporate RWE as a companion to clinical trial data to aid in regulatory decision making. The FDA will release its draft RWE guidance before the end of 2020.
About Aetion
Aetion is a health care technology company that delivers real-world evidence for life sciences companies, payers, at-risk providers, and regulatory agencies. The Aetion Evidence Platform analyzes data from the real world to produce transparent, rapid, and scientifically validated answers on treatments, costs, and outcomes. Founded by Harvard Medical School faculty with decades of experience in epidemiology and health outcomes research, Aetion informs health care's most critical decisions — what works best, for whom, and when — to guide treatment development, commercialization, and payment innovation into health care's modern era. Aetion is based in New York City, and backed by investors including New Enterprise Associates (NEA), Flare Capital Partners, Lakestar, Town Hall Ventures, McKesson Ventures, Sanofi Ventures, Amgen Ventures, UCB, and Horizon Health Services, Inc. Learn more at aetion.com, and follow us at @aetioninc.
About Sanofi
Sanofi is dedicated to supporting people through their health challenges. We are a global biopharmaceutical company focused on human health. We prevent illness with vaccines, provide innovative treatments to fight pain and ease suffering. We stand by the few who suffer from rare diseases and the millions with long-term chronic conditions.
With more than 100,000 people in 100 countries, Sanofi is transforming scientific innovation into healthcare solutions around the globe.
Sanofi, Empowering Life
Sanofi Media Relations Contact
Anna Robinson
Tel.: +33 (0)1 53 77 46 46
mr@sanofi.com
Aetion Media Relations Contact
202.792.7200
press@aetion.com
Sanofi Investor Relations Contact
George Grofik
Tel.: +33 (0)1 53 77 45 45
ir@sanofi.com
Sanofi Forward-Looking Statements
This press release contains forward-looking statements as defined in the Private Securities Litigation Reform Act of 1995, as amended. Forward-looking statements are statements that are not historical facts. These statements include projections and estimates and their underlying assumptions, statements regarding plans, objectives, intentions and expectations with respect to future financial results, events, operations, services, product development and potential, and statements regarding future performance. Forward-looking statements are generally identified by the words “expects”, “anticipates”, “believes”, “intends”, “estimates”, “plans” and similar expressions. Although Sanofi’s management believes that the expectations reflected in such forward- looking statements are reasonable, investors are cautioned that forward-looking information and statements are subject to various risks and uncertainties, many of which are difficult to predict and generally beyond the control of Sanofi, that could cause actual results and developments to differ materially from those expressed in, or implied or projected by, the forward-looking information and statements. These risks and uncertainties include among other things, the uncertainties inherent in research and development, future clinical data and analysis, including post marketing, decisions by regulatory authorities, such as the FDA or the EMA, regarding whether and when to approve any drug, device or biological application that may be filed for any such product candidates as well as their decisions regarding labelling and other matters that could affect the availability or commercial potential of such product candidates, the absence of guarantee that the product candidates if approved will be commercially successful, the future approval and commercial success of therapeutic alternatives, Sanofi’s ability to benefit from external growth opportunities, to complete related transactions and/or obtain regulatory clearances, risks associated with intellectual property and any related pending or future litigation and the ultimate outcome of such litigation, trends in exchange rates and prevailing interest rates, volatile economic conditions, the impact of cost containment initiatives and subsequent changes thereto, the average number of shares outstanding as well as those discussed or identified in the public filings with the SEC and the AMF made by Sanofi, including those listed under “Risk Factors” and “Cautionary Statement Regarding Forward-Looking Statements” in Sanofi’s annual report on Form 20-F for the year ended December 31, 2018. Other than as required by applicable law, Sanofi does not undertake any obligation to update or revise any
forward-looking information or statements.

YTX-7739 represents a novel, first-in-class, potentially disease-modifying therapy
Data from Phase 1 study expected in the first quarter of 2020
CAMBRIDGE, Mass. – October 7, 2019 – Yumanity Therapeutics, a company focused on protecting the vitality of the mind by discovering and developing transformative brain-penetrating small molecule drugs to treat neurodegenerative diseases, today announced that the first subject cohort has been dosed in a Phase 1 clinical trial evaluating the safety and tolerability of YTX-7739 in healthy volunteers. YTX-7739, the company’s lead investigational therapy, is designed to inhibit Stearoyl-CoA-Desaturase (SCD), a validated biologic target that has recently shown potential in neurodegenerative diseases by protecting cells from a-synuclein toxicity, a major driver of Parkinson’s disease.
“Developing effective therapies for patients with devastating neurodegenerative diseases has been challenging because too few hypotheses and novel targets have been explored,” said Kenneth Rhodes, Ph.D., chief scientific officer at Yumanity Therapeutics. “We advanced YTX-7739, an orally-active SCD inhibitor, into clinical development because of recent evidence established at Yumanity Therapeutics demonstrating its promise to protect cells from a-synuclein toxicity. We look forward to fully characterizing the potential clinical use of YTX-7739, which is clearly differentiated from currently available Parkinson’s disease therapies that only address the symptoms, not the underlying causes.”
The double-blind, placebo-controlled, dose-escalation, crossover study is intended to evaluate the safety, tolerability and pharmacokinetics of single ascending doses of YTX-7739 in adult healthy volunteers. A second study, exploring multiple ascending doses in adult healthy volunteers and patients with Parkinson’s, will follow. Approximately 40 participants will be enrolled in this Phase 1 single ascending dose study. Following completion of the Phase 1 studies, Yumanity Therapeutics expects to advance YTX-7739 into a Phase 1b proof-of-concept clinical trial in the second half of 2020.
“Since Yumanity Therapeutics’ inception, our goal has been to uncover novel pathways and targets to tackle significant medical challenges,” said Richard Peters, M.D., Ph.D., chief executive officer of Yumanity Therapeutics. “Moving from target identification of SCD to initial clinical development of YTX-7739 in just three years is a testament to the enormous potential of our discovery platform to reproducibly identify previously unexplored biology and new, druggable targets that have the potential to protect cells from neurodegeneration. This Phase 1 trial will provide important validation for the broad application of our technology to help address arguably the most important therapeutic challenges of our time.”
About YTX-7739
YTX-7739 is Yumanity Therapeutics’ proprietary lead investigational therapy designed to penetrate the blood-brain barrier and inhibit the activity of a novel target that plays an important and previously unrecognized role in the neurotoxicity caused by the a-synuclein protein, a major driver of Parkinson’s disease and related neurodegenerative disorders. Misfolding and aggregation of the a-synuclein protein triggers a cascade of events, ultimately resulting in neurotoxicity and the subsequent disorders in movement and cognition that affect patients living with these diseases. YTX-7739 has been shown to inhibit many of the key aspects of a-synuclein toxicity and the company is assessing its potential utility in Parkinson’s disease.
About Parkinson’s Disease
Parkinson’s disease is a progressive neurological disorder that affects the central nervous system and impacts both motor and non-motor functions. It is one of the most common age-related neurodegenerative diseases, affecting an estimated 0.5 to 1 percent of people 65 to 69 years of age, rising to 1 to 3 percent of the population over the age of 80.1 Symptom severity and disease progression differ between individuals, but typically include slowness of movement (bradykinesia), trembling in the extremities (tremors), stiffness (rigidity), cognitive or behavioral abnormalities, sleep disturbances and sensory dysfunction.2 There is no laboratory or blood test for Parkinson’s disease, so diagnosis is made based on clinical observation.3 Currently, there is no cure and available treatments only address the symptoms of Parkinson’s disease, not the underlying causes.
About Yumanity Therapeutics
Yumanity Therapeutics is transforming drug discovery for neurodegenerative diseases caused by protein misfolding. Formed in 2014 by renowned biotech industry leader, Tony Coles, M.D., and protein folding science pioneer, Susan Lindquist, Ph.D., the company is focused on discovering disease-modifying therapies for patients with Parkinson’s disease and related disorders, amyotrophic lateral sclerosis (ALS), and Alzheimer’s disease. Leveraging its proprietary discovery engine, Yumanity Therapeutics’ innovative new approach to drug discovery and development concentrates on reversing the cellular phenotypes and disease pathologies caused by protein misfolding. For more information, please visit yumanity.com.
# # #
Media Contact:
Rachel Eides
W2O pure
reides@purecommunications.com
Investor Relations Contact:
Caroline Corner, Ph.D.
Westwicke
caroline.corner@westwicke.com
- Two-component virus-like particle (VLP) technology developed at the Institute for Protein Design exclusively licensed from the University of Washington for a variety of infectious disease indications
- License secured from NIH for DS-Cav1 (clinically validated prefusion F antigen) for RSV indication
SEATTLE, October 3, 2019 – Icosavax, Inc. today announced the launch of the company with a $51 million Series A financing, led by Qiming Venture Partners USA and joined by Adams Street Partners, Sanofi Ventures and NanoDimension, with continuing support from its seed investors. The company was founded on computationally designed self-assembling virus-like particle (VLP) technology developed at the Institute for Protein Design (IPD) at University of Washington School of Medicine (Cell 2019, Preview). The proceeds of the financing will be used to advance the company’s first vaccine candidate, IVX-121, for respiratory syncytial virus (RSV) for older adults through Phase 1b clinical studies. Icosavax also announced today its leadership team, board of directors and key scientific advisors.
“Working closely with the Institute for Protein Design, we started Icosavax with seed funding from its philanthropic supporters,” said Adam Simpson, Chief Executive Officer of Icosavax. “This support allowed us to assemble a world-class team and to help translate the scientific insights from IPD into IVX-121, our lead candidate for RSV. We are thrilled with the quality of the investor syndicate we have built who provide both significant expertise and financial support to enable advancement of IVX-121 into the clinic and the application of our VLP technology to a whole class of vaccine targets with significant unmet medical needs.”
VLPs enable high-density, multivalent display of antigens in a manner that closely resembles viruses, with an important difference. VLPs contain no genetic material, so they are non-infectious and can provide a safer alternative to live-attenuated or inactivated vaccines. Naturally occurring VLPs have delivered successful vaccines, including Gardasil® and Cervarix® against human papillomavirus (HPV) and Engerix-B® and Recombivax HB® against Hepatitis B. However, VLPs have been difficult to use for the display of complex heterologous antigens, like in the case of RSV.
Mark McDade, who is managing partner of Qiming Venture Partners USA and was previously a founder, COO and director of Corixa Corporation (sold to GSK), said “We were extremely impressed with this novel approach using computational protein design to create VLP-based vaccines that have improved efficacy and are simple to manufacture. Our investment in Icosavax supports the value of science and technology to improve public health and our belief that preventing infection is preferable to treating illness.”
“Icosavax’s vaccine technology solves the problem of constructing and manufacturing VLPs displaying complex antigens by utilizing computationally designed proteins that separate the folding of individual protein subunits from the assembly of the final macromolecular structure. The individual proteins are expressed and purified using traditional recombinant technologies, and then self-assemble into VLPs when mixed together,” said Icosavax co-founder Neil King, Ph.D. VLPs are known to induce superior immunological responses compared to traditional soluble antigens, eliciting protective immune responses while reducing the need for strong adjuvants, which in some instances have been associated with side effects.
The company’s RSV vaccine candidate, IVX-121, incorporates a stabilized prefusion F antigen licensed from NIAID/NIH (DS-Cav1; Science 2019). Extensive preclinical studies conducted at IPD and Icosavax suggest that IVX-121 could increase the protective immunogenicity of RSV F compared to the DS-Cav1 antigen alone.
Tadataka (Tachi) Yamada, M.D., chair of Icosavax’s board of directors explained, “RSV can cause a life-threatening respiratory infection with consequences as severe as those associated with influenza in older adults. An effective RSV vaccine could have an impact on the lives of millions of people around the world.”
Tachi previously served as chief medical and scientific officer of Takeda Pharmaceuticals, where he was responsible for the acquisitions of Inviragen (dengue vaccine) and LigoCyte (norovirus vaccine). The board of directors also includes Terry Gould, a partner and head of Growth Equity Investments, Adams Street Partners; Jason Hafler, the U.S. Head of Investments at Sanofi Ventures; Eric Moessinger, a partner of NanoDimension, Mark McDade, and Adam Simpson.
Simpson added, “We are pleased to announce our leadership team who has extensive experience in successful vaccine development and our board of directors and key advisors, which include leaders in computational protein design and global vaccine programs, experts in successfully advancing novel technologies to vaccine products, and entrepreneurs who have helped companies develop and create commercially valuable medical products that benefit human health.”
The founding leadership team of Icosavax:
- Adam Simpson is the chief executive officer (CEO). He is also the CEO of PvP Biologics, another spinout from IPD. Previously he served as president and chief operating officer (COO) of Cypher Genomics (sold to Human Longevity), and chief business officer, Meritage Pharma (sold to Shire).
- Doug Holtzman, Ph.D., MPH is the chief scientific officer. Previously, he served as vice president, Discovery, Takeda, and deputy director, Childhood Pneumonia, Bill & Melinda Gates Foundation. Prior to his work in global health he worked in a number of innovative early-stage biotechnology companies, including Millennium Pharmaceuticals (sold to Takeda) and Ironwood.
- Niranjan Kanesa-thasan, M.D., MTMH, FIDSA, FASTMH is the chief medical officer and has contributed to the development of seven licensed vaccines. Previously, he served as the clinical franchise head, GSK Vaccines; chief medical officer, Americas, Novartis Vaccines; and vice president, Medical Affairs and Pharmacovigilance, Acambis (sold to Sanofi Pasteur).
- Charles Richardson, Ph.D. is the senior vice president, Technical Operations. Previously, he served as global head, CMC, Takeda Vaccines; senior vice president, R&D, LigoCyte (sold to Takeda); vice president, Manufacturing, Corixa (sold to GSK); and vice president, Pharma Discovery, RibiImmunoChem (sold to Corixa).
Icosavax’s founding scientists, scientific advisory board (SAB) members, and key advisors are:
- Neil King, Ph.D., is a co-founder of Icosavax, inventor of the computationally designed VLP technology, and chair of the SAB. He is a researcher at the Institute for Protein Design and an assistant professor of biochemistry at the UW School of Medicine.
- David Baker, Ph.D., is a co-founder of Icosavax. He is the director of the Institute for Protein Design, an endowed professor of biochemistry at the University of Washington School of Medicine, and a Howard Hughes Medical Institute (HHMI) investigator.
- Ralf Clemens, M.D., is a development advisor and SAB member and a leading expert in vaccinology with more than 30 years of experience in global vaccine development at Takeda, Novartis, and GSK. Ralf developed and brought to licensure more than 25 different vaccines and has published extensively on vaccines and public health.
- Christian Mandl, M.D., Ph.D., is a SAB member and has an accomplished academic career in molecular and clinical virology. Previously, he served as global head of Research, Early and Exploratory Clinical Development at Novartis Vaccines, leading more than 300 discovery and clinical researchers in the development of a broad range of viral and bacterial vaccines, adjuvants, and delivery platforms.
- Jean-Paul Prieels, Ph.D., is a SAB member and his career spans from basic research to process and product development. He previously served as a senior vice president of Research and Development at GlaxoSmithKline Biologicals (now GSK Vaccines). He was instrumental in developing several commercially available vaccines, such as rotavirus, human papilloma virus (HPV), pneumococcal conjugates, and others.
- Barney S. Graham, M.D., Ph.D., is a SAB member, and inventor of DS-Cav1, the clinically validated RSV F antigen incorporated into IVX-121. He is an immunologist, virologist, and clinical trials physician. He was one of the founding investigators for the National Institute of Allergy and Infectious Diseases (NIAID) Vaccine Research Center (VRC) at the National Institutes of Health (NIH), where he is now the deputy director and chief of the Viral Pathogenesis Laboratory and oversees the advanced development of VRC candidate vaccine products.
Icosavax’s in-licensed computationally designed VLP technology is a product of the IPD’s Translational Investigator Program, which enables entrepreneurial researchers to turn their first working versions of designed proteins into commercially viable assets. The license to this technology was negotiated with CoMotion at the University of Washington, UW’s collaborative innovation hub.
About Icosavax
Icosavax is focused on developing safe and effective vaccines against infectious diseases that address important unmet medical needs and reduce healthcare costs. The company was founded on breakthrough computationally-designed virus like particle technology, exclusively licensed for a variety of infectious disease indications from the Institute for Protein Design at the University of Washington. Icosavax is located in Seattle. For more information, please visit www.icosavax.com.
Media Contact:
Jessica Yingling, Ph.D.
Little Dog Communications Inc.
jessica@litldog.com
+1.858.344.8091
Funding to Advance Development of Drug Candidates Targeting
Heparin-Induced Thrombocytopenia and Type 1 Diabetes
FREDERICK, MD – September 5, 2019 – Veralox Therapeutics (“Veralox”), a preclinical stage company focused on accelerating the development of first-in-class therapeutics for unmet medical needs, today announced it has raised a $5.4 million seed funding round. The funding was led by the JDRF T1D Fund, Sanofi Ventures, and the VTC Innovation and VTC Seed Fund, with participation from the Maryland Momentum Fund, the University of Vermont Health Network and TEDCO.
Veralox is headquartered in Maryland and founded in 2017 by Drs. Jeffrey Strovel, David Maloney, and Matthew Boxer, three industry veterans with a track record of successful drug discovery and development. The company is currently developing therapeutics targeting 12- lipoxygenase (12-LOX). The new funding will be used to support the pre-clinical development of therapeutics to block the 12-LOX enzyme for several indications, including the rare hematological indication heparin-induced thrombocytopenia and thrombosis (HITT) and type 1 diabetes (T1D). Veralox’s lead candidate, VLX-1005, was discovered and developed in collaboration with University of California Santa Cruz, Thomas Jefferson University, NIH, and Eastern Virginia Medical School.
“We are excited to fuel our mission to advance the development of therapeutics for immunemediated disorders such as HITT and T1D,” said Jeffrey Strovel, CEO of Veralox Therapeutics. “With the support of the T1D Fund, Sanofi Ventures, and our other investors, we are well positioned to broaden the potential indications for our technology while ensuring sufficient capital to prepare for clinical studies in patients.”
“Beta cell therapies are a key strategic area of focus for the T1D Fund and we believe Veralox’s innovative approach to inhibiting beta cell stress has the potential to create a first-in-class drug targeting a novel pathway to address type 1 diabetes,” said Katie Ellias, Managing Director at the JDRF T1D Fund. “JDRF was an early supporter of work on this pathway, and we look forward to supporting the company and collaborating with Sanofi and the other investors to accelerate the development of this promising therapy.”
“Veralox fits within our strategic investing mandate and we are excited by the potential for 12-LOX inhibition in both T1D and HITT. We see the potential to impact patients across the globe in these diseases with high morbidity and mortality,” said Daniel Mansuri from Sanofi Ventures.
Both Daniel Mansuri and Katie Ellias will join the Board of Directors.
Veralox is pioneering the development of inhibitors of 12-LOX, which plays key a role in platelet activation and mediation of cellular stress signaling. Halting the immune driven activation of platelets reduces blood clot formation, offering the promise of an orphan hematological indication for the immune-mediated coagulation disorder known as heparin-induced thrombocytopenia and thrombosis (HITT). In T1D, beta cell stress leads to cell dysfunction and death. By keeping beta cells healthy and functional, Veralox’s inhibitor may halt T1D disease progression and has the potential to be used at all stages of the disease.
###
About Veralox Therapeutics
VERALOX Therapeutics Inc. (https://veralox.com/) is developing first-in-class therapeutics that target the underlying pathologies of thrombosis and type 1 diabetes. The company’s lead candidate, VLX-1005, will be developed initially to treat patients with heparin-induced thrombocytopenia (HIT) and HIT with thrombosis (HITT). Second generation therapeutic products are under development for type 1 diabetes (T1D) as well as other immune-mediated and inflammatory diseases.
JDRF T1D Fund
The JDRF T1D Fund (https://t1dfund.org/) is a venture philanthropy fund accelerating lifechanging solutions to cure, prevent and treat type 1 diabetes (T1D) through catalytic commercial investments. Through its investments in partnership with private capital, including venture capital, corporations and foundations, the T1D Fund seeks to attract the private investment necessary to advance drugs, devices, diagnostics, and vaccines into the hands of
those living with T1D. The T1D Fund invests in areas strategically aligned with JDRF, the leading global organization funding T1D research, with an exclusive focus on supporting the best commercial opportunities. The T1D Fund will reinvest any realized gains into new investments to further its mission.
Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi. Sanofi Ventures invests in earlystage biotech and digital health companies with innovative ideas and transformative new products and technologies of strategic interest to Sanofi. Among these areas are rare diseases, vaccines, potential cures in other core areas of Sanofi’s business footprint, and digital health solutions. For more information, visit www.sanofiventures.com.
Media Contacts:
Contact@Veralox.com
301-360-3502
JDRF T1D Fund
Scott Lessne
Stanton
slessne@stantonprm.com
646-502-3569
NEW YORK – September 4, 2019 - Click Therapeutics, Inc. (“Click”), a leader in Digital Therapeutics™ solutions as prescription medical treatments, today announced the appointment of Ross J. Muken as Chief Financial Officer. Mr. Muken brings to Click over 18 years of Wall Street experience and a distinguished track record of success, having been broadly recognized as a thought leader across the healthcare sector. Mr. Muken joins the Click team from Evercore ISI, where he was a Senior Managing Director and Partner.
“We are thrilled to welcome Ross to the leadership team,” said David Benshoof Klein, co-founder and CEO of Click. “Ross has had unrivaled insight into our industry since inception. His deep expertise across the healthcare continuum, strong relationships within the community, and wealth of experience will help us to capitalize on our leading position as we transition toward the next stage of rapid growth.”
“I’ve had the privilege of covering a number of disruptive technology companies in the healthcare space throughout my career, and Click is the most exciting opportunity I have come across to-date, with a clear path to becoming the leader in the digital therapeutics space,” said Ross J. Muken. “I am extremely impressed by the tremendous progress that Click has made in the past year and am pleased to join Click at this pivotal time as it is advancing several first-line digital therapeutics into the market. I look forward to leveraging my experience to support Click’s future growth and financial strategy as it builds upon the industry’s most advanced digital therapeutics and patient engagement platform.”
Ross J. Muken was previously Partner, Senior Managing Director and Head of the Healthcare Services & Technology Research Team at Evercore ISI, widely recognized as the #1 independent equity research franchise on Wall Street. He provided thoughtful insights on over 50 companies across the Healthcare Technology & Distribution, Life Science Tools & Diagnostics, Managed Care & Facilities and Medical Supplies & Devices sub-sectors. Ranked amongst the top analysts by Institutional Investor over the past decade, Mr. Muken is well-known for his proprietary fundamental research and highly accurate financial modeling as well as his work with emerging and disruptive technologies across the healthcare continuum. Prior to joining Evercore ISI, Mr. Muken served as a Managing Director at Deutsche Bank Securities, where he helped build the healthcare platform into a top franchise at the company. Previously, Mr. Muken spent time in the Equity Research division of Thomas Weisel Partners and began his career as an M&A Investment Banker at Bank of America Securities. Mr. Muken earned his B.S. in Business Administration, magna cum laude, at Boston University.
About Click Therapeutics
Click Therapeutics, Inc. develops and commercializes software as prescription medical treatments for people with unmet medical needs. Through cognitive and neurobehavioral mechanisms, Click’s Digital Therapeutics™ enable change within individuals, and are designed to be used independently or in conjunction with biomedical treatments. The Clickometrics® adaptive data science platform continuously personalizes user experience to optimize engagement and outcomes. Following a groundbreaking clinical trial, Click’s industry-leading smoking cessation program is available nationwide through a wide variety of payers, providers, and employers. Click’s lead prescription program is entering into a multi-center, randomized, controlled, parallel-group, phase III FDA registration trial for the treatment of Major Depressive Disorder in adults. For more information, visit ClickTherapeutics.com.
101 Avenue of the Americas, 8th Floor
New York, NY 10013
www.clicktherapeutics.com
# # #
Company Contact
Sarah Jackson
Chief of Staff
sarah@clicktherapeutics.com
Media Contacts
Karen Sharma
ksharma@macbiocom.com
781-235-3060
Digital Care Leader’s Integrated Program Supports Individuals Dealing with Chronic
Disease, Mental Health Issues
San Francisco, CA; (June 26, 2019) - Omada Health today announced a $73 million round of funding led by Wellington Management Company LLP. Wellington Management is an independent investment firm with more than $1 trillion of client assets under management. Omada will use the funding to fuel the continued expansion of its digital care program, including support for those with type 2 diabetes and hypertension, as well as those dealing with anxiety and depression.
Omada works with more than 600 employers and health plans across all 50 states, delivering an integrated experience that adapts to participants health needs, and personalizes their journeys to create the best health outcomes. Omada’s dedicated coaches guide each individual throughout the program, achieving unparalleled engagement and sustainable behavior change. Omada’s proprietary coaching platform enables them to deliver personalized interventions at scale. The company proved the model for digital care as the nation’s largest CDC-recognized provider of the National Diabetes Prevention Program, and has now launched programs over the last twelve months for diabetes self-management, as well as anxiety and depression, to holistically address individuals’ health needs.
“Ten years from now, the most engaging and utilized healthcare provider in the U.S. will be digital. Omada is poised to be that provider, as we inspire and engage the more than 100 million patients who need additional care and support between physician visits,” said Omada Health CEO and co-founder Sean Duffy. “Today’s announcement -- and the incredible roster of investors participating in this round of fundraising -- will deepen our collaboration with health plans, employers, and health systems, and accelerate the development of our truly personalized program that helps our participants build patterns that spark lifelong health.”
“Omada has become a category-defining company in digital healthcare,” added Vijay Pande, General Partner at Andreessen Horowitz. “Since our first investment, the team’s vision, rigorous clinical focus, and intelligent application of sustainable behavior change across multiple conditions has raised the bar for what health plan buyers, and individuals, should expect from digital health.”
“Through our collaboration with Omada, we’re making it simple and convenient for our customers to take control of their health and well-being. By harnessing the power of personal coaching, peer support, digital engagement and personal accountability, we can better inspire and support people to achieve their individual health goals and prevent chronic health conditions,” said Joan Harvey, Cigna’s Senior Vice president of Consumer Health Engagement and Behavioral Health. “We’re excited to deepen our relationship with Omada, and further integrate its innovative program into our full suite of data-driven health services.”
Omada has published 11 peer-reviewed studies, and is currently running the largest-ever randomized controlled trial of digital diabetes prevention. The company’s approach is proven to deliver lasting clinical outcomes, as well as significant medical cost savings, across diverse populations. Omada’s program drives unprecedented levels of meaningful engagement by participants, directly leading to sustained improvements in health.
Also participating in the round were previous investors Cigna Ventures, Andreessen Horowitz, U.S. Venture Partners, Norwest Venture Partners, Kaiser Permanente Ventures, Sanofi Ventures, Civilization Ventures, and Providence Ventures.
About Omada
Omada is a digital care program that empowers people to achieve their health goals through sustainable lifestyle change. Working primarily through health plans, employers, and integrated health systems, the company delivers personalized interventions for individuals at risk for, or dealing with, type 2 diabetes and hypertension, as well as anxiety and depression. Combining data-powered human coaching, connected devices, proprietary technology platform, and curriculum tailored to an individual's specific conditions and circumstances, Omada has enrolled more than 250,000 participants to date. Omada partners include Cigna, Kaiser Permanente, Health Care Services Corporation (HCSC), Blue Cross Blue Shield Minnesota, and other leading health plans. For additional information, please visit www.omadahealth.com .
-Multi-year collaboration leveraging Kymera’s proprietary targeted protein degradation platform to develop novel medicines-
-Kymera to receive $70 million upfront, including equity investment, and potential additional milestone and royalty payments for up to six programs in the collaboration-
BOSTON, MA and CAMBRIDGE, MA – May 15, 2019 – Vertex Pharmaceuticals Incorporated (NASDAQ:VRTX) and Kymera Therapeutics today announced that the two companies have entered into a four-year strategic research and development collaboration to advance small molecule protein degraders against multiple targets. The collaboration will leverage Kymera’s expertise in targeted protein degradation and its proprietary PegasusTM drug discovery platform and Vertex’s scientific, clinical, and regulatory capabilities to accelerate the development of first-in-class medicines for people with serious diseases.
“This collaboration with Kymera will enhance our drug discovery capabilities and support our strategy of investing in scientific innovation to develop transformative medicines for serious diseases,” said Mark Bunnage, Senior Vice President and Site Head, Boston Research at Vertex. “We’ve been impressed by the Kymera team’s depth of knowledge in the field and compelling technology platform, and are excited to bring our research and development expertise to this promising new therapeutic modality.”
“We are thrilled to partner with Vertex to combine their deep understanding of human biology and genetics with Kymera’s state-of-the art protein degrader platform,” said Laurent Audoly, PhD, president and CEO, Kymera Therapeutics. “This strategic partnership will broaden the application of targeted protein degradation to address serious diseases beyond cancer with limited or no treatment options. This fits perfectly with Kymera’s vision to build a platform that is disease agnostic and delivers the broadest possible impact.”
About the Collaboration
Under the terms of the four-year agreement, Vertex will pay Kymera $70 million upfront including an equity investment in the company. Kymera will conduct research activities in multiple targets under the collaboration. Upon designation of a clinical development candidate, Vertex has the option to exclusively license molecules against the designated target. Kymera is also eligible to receive more than $1 billion in potential payments based upon the successful achievement of specified research, development, regulatory, and commercial milestones for up to six programs optioned as part of the collaboration. In addition, Vertex will pay tiered royalties on future net sales on any products that may result from this collaboration.
About Kymera Therapeutics
Kymera Therapeutics is a biotechnology company pioneering a transformative new approach to treating previously untreatable diseases. The company is advancing the field of targeted protein degradation, accessing the body’s innate protein recycling machinery to degrade dysregulated, disease-causing proteins. Powered by Pegasus™, a game-changing integrated degradation platform, Kymera is accelerating drug discovery with an unmatched ability to target and degrade the most intractable of proteins, and advance new treatment options for patients. For more information visit, www.kymeratx.com.
About Pegasus™
Pegasus™ is Kymera Therapeutics’ proprietary protein degradation platform, created by its team of experienced drug hunters to improve the effectiveness of targeted protein degradation and generate a pipeline of novel therapeutics for previously undruggable diseases. The platform consists of informatics driven target identification, novel E3 ligases, proprietary ternary complex predictive modeling capabilities and degradation tools.
About Vertex
Vertex is a global biotechnology company that invests in scientific innovation to create transformative medicines for people with serious and life-threatening diseases. In addition to clinical development programs in CF, Vertex has more than a dozen ongoing research programs focused on the underlying mechanisms of other serious diseases.
Founded in 1989 in Cambridge, Mass., Vertex’s headquarters is now located in Boston’s Innovation District. Today, the company has research and development sites and commercial offices in the United States, Europe, Canada, Australia and Latin America. Vertex is consistently recognized as one of the industry’s top places to work, including being named to Science magazine’s Top Employers in the life sciences ranking for nine years in a row.
For additional information and the latest updates from the company, please visit www.vrtx.com.
Special Note Regarding Forward-Looking Statements
This press release contains forward-looking statements as defined in the Private Securities Litigation Reform Act of 1995, including, without limitation, Dr. Bunnage’s statements in the second paragraph of the press release, Dr. Audoly’s statements in the third paragraph of the press release, and statements regarding future activities of the parties pursuant to the collaboration. While Vertex believes the forward-looking statements contained in this press release are accurate, these forward-looking statements represent Vertex’s beliefs only as of the date of this press release and there are a number of factors that could cause actual events or results to differ materially from those indicated by such forward-looking statements. Those risks and uncertainties include, among other things, Vertex may not realize the anticipated benefits of the collaboration, and the other risks listed under Risk Factors in Vertex’s annual report and quarterly reports filed with the Securities and Exchange Commission and available through the company’s website at www.vrtx.com. Vertex disclaims any obligation to update the information contained in this press release as new information becomes available.
Vertex Contacts:
Investors:
Michael Partridge, 617.341.6108
Eric Rojas, 617.961.7205
Zach Barber, 617.341.6470
Media:
617.341.6992
mediainfo@vrtx.com
Kymera Contact:
Lissette Steele
Verge Scientific Communications
lsteele@vergescientific.c
Sanofi, UCB, McKesson Ventures, and Horizon Health Services Endorse Aetion's Vision for Real-World Evidence
NEW YORK, Feb. 5, 2019 /PRNewswire/ -- Aetion, the health care technology company that delivers the real-world analytics and evidence platform needed to engage in value-based care, announced it has attracted strategic investment from global leaders in biopharma and health care services, including Sanofi, UCB, McKesson Ventures, and Horizon Health Services, Inc., a Blue Cross Blue Shield of New Jersey company — four new strategic shareholders that join Amgen Ventures, an earlier investor. Existing venture capital investors also participated in this round, including NEA, Flare Capital, and Lakestar.
The latest strategic capital infusion of $27 million reflects Aetion's substantialgrowth in customer adoption over the past year. Coupled with its prior funding in early 2018, the investment caps the total Series B funding at $63 million. This brings Aetion's total funding to $77 million since its 2015 launch. Investment funds will be applied to strengthen the Aetion Evidence Platform™, extending its reach in therapeutic area-related intelligence, outcomes-based contracting for payers, and advancing the acceptance of standards for real-world evidence around the globe.
"The time for real-world evidence is now," said Carolyn Magill, Chief Executive Officer of Aetion. "We're entering a new era in which nearly the entire health care ecosystem — from biopharma and regulators to payers and technology companies— recognizes the importance of using real-world evidence to reduce the time and cost to bring new therapies to market. This funding demonstrates that the industry's leaders, who are using our technology to drive health care's most critical decisions, view Aetion as a trusted partner vital to their long-term success."
For Aetion, strategic backing from a sweep of biopharma, health care services, and payer organizations with investment capital reinforces its position as an unbiased thought-leader at the forefront of industry adoption of real-world evidence, and underscores growing confidence among its partners and clients in the Aetion Evidence Platform as the analytics tool of record.
"We are at an inflection point, in which the convergence of massive analytical advances and enormous amounts of new data are transforming what's possible in health care," said Ameet Nathwani, M.D., Chief Medical Officer at Sanofi. "Aetion is ideally positioned to lead this shift, given the power of its technology and extensive experience with biopharma leaders and the FDA. Sanofi is excited to join as a strategic investor as the company helps set the global standards for real-world evidence."
"Aetion's abilities represent the leading edge of member-centric, value-based care," said Bill Georges, Chief Strategy Officer at Horizon Healthcare Services, Inc. "Horizon has successfully deployed Aetion's analytics and leveraged real-world evidence to determine which treatments are most effective for specific patient populations with diabetes. Horizon shares this data with our members' doctors to help them deliver more effective treatment regimens that work to improve outcomes for our members and reduce their overall cost of care. We are thrilled to be collaborating with Aetion as a strategic investor and excited to expand the number of Horizon members who will benefit directly from Aetion's innovation."
Aetion is coming off a year of strong growth, and this funding continues the company's momentum. Today, the Aetion Evidence Platform is used by eight of the top 15 global biopharma firms, leading payers, academic institutions, the U.S. Food and Drug Administration (FDA) and other major international regulatory bodies. Since 2017, it has seen a 100 percent customer renewal rate.
"Aetion shares our ambition to transform the future for people living with severe diseases, and our passion for science-driven approaches that serve this goal," said Iris Loew-Friedrich, M.D., Ph.D., chief medical officer at UCB. "Aetion's transparent
evidence serves as a bridge between UCB and its payers, enabling us to apply realworld evidence to engage them in new ways. We're thrilled to build upon our strong relationship with Aetion as a strategic investor."
Also in 2018, Aetion began a partnership with the FDA to recreate 30 randomized clinical trials through real-world evidence, to demonstrate its value as an accelerant to drug approval and access decisions. The three-year study, named DUPLICATE, is referenced in the FDA's recently released Framework for its Real-World Evidence Program, which applies across its drug and biologic review programs — yet another clear signal that real-world evidence will modernize how treatments are developed, approved, and commercialized.
"Aetion is poised to be the partner of choice for biopharma companies, payers and regulators looking to leverage real-world evidence given its unique relationships across the health care ecosystem and its expertise in protocols and treatment based analytics," said Tom Rodgers, SVP and Managing Director at McKesson Ventures.
To learn more, visit Aetion's website and subscribe to its newsletter at www.aetion.com.
About Aetion
Aetion is a health care technology company that delivers real-world evidence for life sciences, payers, providers, and regulatory agencies. The Aetion Evidence Platform analyzes data from the real world to produce transparent, rapid, and scientifically validated answers on treatments, costs, and outcomes. Founded by Harvard Medical School faculty members with decades of experience in epidemiology and health outcomes research, Aetion informs health care's most critical decisions — what works best, for whom, and when — to guide treatment development, commercialization, and payment innovation into health care's modern era.
Aetion is based in New York City, and backed by investors including New Enterprise Associates (NEA), Flare Capital Partners, Lakestar, Town Hall Ventures, McKesson Ventures, Sanofi Ventures, Amgen Ventures, UCB, and Horizon Health Services, Inc. Learn more at aetion.com, and follow us at @aetioninc.
SOURCE Aetion
Related Links
https://www.aetion.com
Safety Data and Potential Clinical Efficacy Signal Expected Mid-2019
CAMBRIDGE, Mass.--(BUSINESS WIRE)-- Navitor Pharmaceuticals, Inc., the leader in the development of mTORC1-targeted therapeutics designed to help patients live longer and healthier lives, announced today the initiation of Part B of its Phase 1 clinical study with its lead candidate, NV-5138, for treatment-resistant depression (TRD). NV-5138 is a first-in-class, orally-active small molecule that directly activates mTORC1, the gatekeeper of cellular metabolism and renewal, which is suppressed in the brain of people suffering from depression.
“NV-5138’s direct activation of mTORC1 may deliver significant advantages over antidepressants that only indirectly activate this master metabolic control switch, offering the potential for rapid-acting antidepressant benefits without the psychotomimetic side effects and abuse potential observed with many N-methyl-D-aspartic-acid (NMDA) receptor targeted therapeutics,” said Thomas E. Hughes, Ph.D., Chief Executive Officer of Navitor. “The positive results from the single ascending dose portion of our Phase 1 clinical study of NV-5138 in healthy volunteers support advancement into Part B, and we are now evaluating a single dose of the compound in patients suffering with TRD. We look forward to the initial top-line data from this study in the middle of 2019.”
The Phase 1, multicenter, two-part, double-blind, placebo-controlled study is evaluating the safety, tolerability and pharmacokinetics of NV-5138 in up to 88 subjects, including healthy volunteers and patients diagnosed with TRD. In Part A, the single-ascending-dose portion of the study, up to 48 healthy volunteers were randomly assigned to double-blind treatment in six cohorts. In Part B of the study, approximately 40 subjects diagnosed with TRD are being randomized to receive either a single dose of NV-5138 or placebo. Secondary efficacy outcome measures for Part B include standard depression rating and symptomology scores such as the Montgomery-Åsberg Depression Rating Scale (MADRS).
“In multiple standard preclinical models of depressive behavior and cognition, we have shown that a single dose of NV-5138 stimulates mTORC1, enhances protein expression within hours, and increases synaptic growth in key, relevant brain regions, resulting in sustained antidepressant behavioral responses,” stated George P. Vlasuk, Ph.D., President and Chief Scientific Officer of Navitor. “These behavioral changes and increases in synaptogenesis were consistent with the effects of NMDA receptor modulators such as ketamine; however, NV-5138 works through direct, post-synaptic activation of the mTORC1 signaling pathway and may therefore offer the potential for an improved safety and tolerability profile. Part B of our Phase 1 trial will offer important insights on the candidate’s potential in this difficult-to-treat patient population.”
About NV-5138
NV-5138 is an orally bioavailable, small molecule that directly and transiently activates mTORC1, the master modulator of cellular metabolism, which is often suppressed in the brain of patients suffering from depression. NV-5138 binds to and modulates sestrin, a newly discovered cellular sensor protein for the amino acid leucine, a potent natural activator of mTORC1. As opposed to many other organ systems like skeletal muscle, leucine is a poor activator of mTORC1 in the brain since it is principally used as a metabolic precursor for neurotransmitter and protein synthesis. NV-5138 was designed to avoid the metabolic fate of leucine in the brain and thus serves as an effective activator of mTORC1 in this tissue. Results from preclinical models demonstrate that oral administration of NV-5138 produces rapid upregulation of key synaptic proteins, synaptic remodeling in the prefrontal cortex and hippocampus, sustained antidepressant behavioral responses, cognitive improvements and compound-specific spectral power changes, as measured by quantitative electroencephalography (qEEG). Navitor’s strong intellectual property portfolio includes composition of matter patent protection for NV-5138 and related compounds.
About mTORC1
mTORC1, or Complex 1 of the mechanistic target of rapamycin, activity governs the pace and ability of the cell to synthesize protein and other cellular components. Increased mTORC1 activity contributes to a broad array of diseases of aging by increasing protein misfolding and driving cellular stress, inflammation, and fibrosis. In other disease states such as severe depression, inadequate mTORC1 activity contributes to disease pathology by limiting energy utilization and protein synthesis, leading to impaired function. Multiple preclinical studies have shown that mTORC1 activation is required for the efficacy of many rapid-acting antidepressant compounds, including but not limited to modulators of the N-methyl-D-aspartic-acid (NMDA)-mediated signaling pathway like ketamine.1
About Navitor
Navitor Pharmaceuticals, Inc. is the leader in the development of mTORC1-targeted therapeutics designed to help patients live longer and healthier lives. The Company’s proprietary platform enables true modulation of mTORC1, the gatekeeper of cellular metabolism and renewal, with the first-ever absolutely selective mTORC1 inhibition and the unique ability for mTORC1 activation. Navitor’s lead clinical-stage candidate, NV-5138, is a small molecule that directly activates mTORC1 and is being developed for treatment-resistant depression, with additional opportunities in cognition and memory. The Company’s NΛValog program, which provides unprecedented selectivity in mTORC1 inhibition, is initially targeting chronic kidney disease and has broad potential application for age-related diseases. For more information, please visit www.navitorpharma.com.
Approach combines Otsuka’s expertise in developing and commercializing treatments for mental health with Click’s record of discovering and validating digital technologies as prescription medical treatments
SAN FRANCISCO, C.A. and NEW YORK, N.Y. – January 3, 2019 – Otsuka America, Inc., and Click Therapeutics, Inc., announce today that the companies have signed a collaboration agreement to develop and commercialize a prescription digital therapeutic for treatment of Major Depressive Disorder (MDD), with the intent to address unmet medical needs among this patient population and to improve outcomes.
This collaboration will leverage Click’s demonstrated ability to discover and validate a software application and deploy it commercially, with Otsuka’s expertise in developing approved prescription therapies for patients with serious mental illnesses, including Otsuka’s established development and commercialization capabilities. The companies believe digital therapeutics align naturally with psychiatry and have significant potential to transform mental healthcare. Together, the companies aim to bring to market a new offering that will provide a novel treatment for patients with MDD.
Otsuka has agreed to commit capital to fully fund development of Click’s novel mobile application ‘CT-152’ for MDD, and to commercialize this application world-wide upon achievement of regulatory approvals. Otsuka will pay Click up to $10 million in upfront and regulatory milestone payments, along with an estimated $20 million in development funding. An additional $272 million in commercial milestone payments are contingent upon regulatory approvals. In addition, Click will receive tiered, double-digit royalties on global sales of the software and the digital therapeutic applications that result.
“This collaboration signals Otsuka’s commitment to meet patients’ unmet medical needs by developing solutions far beyond medication. Our goal is to deliver evidence-based cognitive therapies to a broader population of patients with MDD than is currently feasible, due to the challenges of a shortage of mental health professionals and limited time for them to conduct cognitive therapy,” said Kabir Nath, president and CEO, Otsuka North America Pharmaceutical Business Division, Otsuka America, Inc. “We are proud to be one of the few pharmaceutical companies that continues to invest in developing medicinal and digital products for the treatment of mental illnesses, and we are doing so by breaking down barriers and collaborating with leading therapeutic technology companies, such as Click, which share our vision.”
According to the World Health Organization, depression is the leading cause of disability worldwide, and is a major contributor to the overall global burden of disease.1 Otsuka and Click believe that new approaches are needed to address this condition, including pioneering ways to use technology and data in support of better patient outcomes.
“Otsuka has established itself as the leading innovator in digital medicine for psychiatry, and we are thrilled to collaborate with their clinical and commercial experts on our CT-152 digital therapeutic for the treatment of depression. This collaboration symbolizes the growing recognition that digital therapeutics are a new category of treatment with the potential to become a routine treatment option for physicians for their patients,” said David Benshoof Klein, Chairman and CEO of Click. “The potential value of these treatments is clear: they provide validated tools that may be an effective treatment option to bring the power and accessibility of digital technologies for the benefit of the patient and physician. This recognition is also shared by regulators helping to build new pre- market review programs uniquely suited for digital therapeutics, while still retaining the traditional level of rigor and scrutiny that clinical studies require.”
About the prescription digital therapeutic for MDD and the agreement between Otsuka and Click:
- CT-152 is a software application (app) that will leverage evidence-based cognitive therapy principles and Click’s patient engagement platform to treat patients either independently or in conjunction with prescribed pharmacotherapies.
- The intent is that the app will be classified as Software as a Medical Device (SaMD) and will fall under the FDA regulatory framework that supports innovation and commercialization of digital tools while protecting patient health.
About Otsuka Pharmaceutical
Otsuka Pharmaceutical is a global healthcare company with the corporate philosophy: “Otsuka-people creating new products for better health worldwide.” Otsuka researches, develops, manufactures and markets innovative products, with a focus on pharmaceutical products for the treatment of diseases and nutraceutical products for the maintenance of everyday health.
In pharmaceuticals, Otsuka is a leader in the challenging area of mental health and has research programs on several under-addressed diseases including tuberculosis, a significant global public health issue. These commitments illustrate how Otsuka is a “big venture” company at heart, applying a youthful spirit of creativity in everything it does.
Otsuka America, Inc. is a subsidiary of Otsuka Pharmaceutical Co., Ltd. headquartered in Tokyo, Japan. Both are part of Otsuka Holdings Co., Ltd. The Otsuka group of companies employed 46,000 people worldwide and had consolidated sales of approximately USD 11.1 billion in 2017.
All Otsuka stories start by taking the road less travelled. Learn more about Otsuka Pharmaceutical Company on its global website at https://www.otsuka.co.jp/en. Learn more about Otsuka in the U.S. at www.otsuka-us.com. Connect with us in the U.S. on Twitter at @OtsukaUS.
About Click Therapeutics
Click Therapeutics, Inc. develops and commercializes software as prescription medical treatments for people with unmet medical needs. Through cognitive and neurobehavioral mechanisms, Click’s Digital Therapeutics™ enable change within individuals, and are designed to be used independently or in conjunction with biomedical treatments. The Clickometrics® adaptive data science platform continuously personalizes user experience to optimize engagement and outcomes. Following a groundbreaking clinical trial, Click’s industry-leading smoking cessation program is available nationwide in the U.S through a wide variety of payers, providers, and employers. Click’s lead prescription program is entering into a multi-center, randomized, controlled, parallel-group, phase III FDA
December 2018 01US18EUC0370
December 2018 01US18EUC0370
registration trial for the treatment of Major Depressive Disorder in adults. For more information, visit ClickTherapeutics.com.
References:
1. World Health Organization. Depression Fact Sheet. Available at: www.who.int/news-room/fact- sheets/detail/depression
Media Contacts
Otsuka In U.S.
Robert Murphy
Corporate Communications
Otsuka America Pharmaceutical, Inc.
robert.murphy@otsuka-us.com
+1 609 249 7262
Otsuka in Japan
Jeffrey Gilbert (Outside the U.S.)
Leader, Pharmaceutical PR
Otsuka Pharmaceutical, Co., Ltd.
gilbert.jeffrey@otsuka.co.jp
+81 3 6361 7379, +81 80 8728 6039
Click Therapeutics
Contact Karen Sharma
ksharma@macbiocom.com
+1 781 235 3060
Series B financing led by 6 Dimensions Capital, Bessemer Venture Partners, and Pfizer Ventures
Financing to enable the advancement of lead program into clinical development and support the progression of additional programs
Cambridge, Mass. (Nov. 13, 2018) – Kymera Therapeutics Inc., a biotechnology company pioneering targeted protein degradation to create breakthrough medicines for patients, announced today that it has completed a $65 million Series B financing. The financing will support the advancement of its lead asset into clinical development, and to progress its therapeutic pipeline in oncology and immunology. The financing was co-led by 6 Dimensions Capital, Bessemer Venture Partners, and Pfizer Ventures with participation by MRL Ventures Fund, Sanofi Ventures, Hatteras Venture Partners, and Aju IB Investment, in addition to Kymera’s Series A investors. The company also announced Wei Li, PhD, of 6 Dimensions Capital, Andrew Hedin of Bessemer Venture Partners, and Elaine Jones, PhD, of Pfizer Ventures, will join Kymera Therapeutics’ Board of Directors.
“We are delighted to have the support of this exceptional group of both new and existing investors and experts that share our vision and excitement on our progress to date,” said Laurent Audoly, PhD, president and CEO, Kymera Therapeutics. “In the last year, we’ve made great strides in building preclinical data packages supporting both the drug-like properties of our assets and their differentiated pharmacology. This rapid advancement has been enabled by our focused investment in Kymera’s industry-leading protein degradation platform and a stellar team of drug hunters and seasoned executives.”
Since announcing its founding in 2016, Kymera Therapeutics has progressed its proprietary Pegasus™ integrated degradation platform, consisting of informatics-driven target identification coupled to ternary complex predictive modeling, new degradation tools, and novel E3 ligases and ligands. Kymera also entered into a two-year discovery collaboration agreement with GSK, leveraging innovations in small molecule-based targeted protein degradation and encoded library technologies.
“This year has been marked by the maturation of our pipeline with applications across diverse target types and disease indications as well as a deeper understanding of the pharmaceutical properties required to develop protein degrading drugs,” said Nello Mainolfi, PhD, co-founder and chief technology officer, Kymera Therapeutics. “These insights have allowed us to refine our drug discovery platform and improve our ability to identify first-in-class drug candidates.”
“Kymera is truly at the forefront of targeted protein degradation, one of the most exciting new areas of drug discovery and medicine,” said Bruce Booth, DPhil, Kymera co-founder, partner at Atlas Venture, and chairman of the board. “With an incredible team, advisors and syndicate and a singular focus on rational drug discovery, the company has quickly emerged as a leader in the field and is well-positioned to realize the potential of this transformational modality to impact patients.”
About Kymera Therapeutics
Kymera Therapeutics is a biotechnology company pioneering a transformative new approach to treating previously untreatable diseases. The company is advancing the field of targeted protein degradation, accessing the body’s innate protein recycling machinery to degrade dysregulated, disease-causing proteins. Powered by Pegasus™, a gamechanging integrated degradation platform, Kymera is accelerating drug discovery with an unmatched ability to target and degrade the most intractable of proteins, and advance new treatment options for patients. For more information visit, www.kymeratx.com.
About Pegasus™
Pegasus™ is Kymera Therapeutics’ proprietary protein degradation platform, created by its team of experienced drug hunters to improve the effectiveness of targeted protein degradation and generate a pipeline of novel therapeutics for previously undruggable diseases. The platform consists of informatics driven target identification, novel E3 ligases, proprietary ternary complex predictive modeling capabilities and degradation tools.
About 6 Dimensions Capital
6 Dimensions Capital is a leading healthcare focused investment firm with an in-depth focus and extensive coverage across China and the United States. It consists of a team of about 40 investment and operation professionals with offices in Shanghai, Hong Kong, Boston, and Palo Alto. The firm currently has US$1.6 billion assets under management through 4 US dollar-denominated and 3 RMB-denominated funds and has cultivated a portfolio of more than 80 companies. For more information, visit www.6dimensionscapital.com.
About Pfizer Ventures
Pfizer Ventures (PV), the venture capital arm of Pfizer Inc. was founded in 2004 and invests for return in areas of current or future strategic interest to Pfizer. PV seeks to remain at the forefront of life science advances, looking to identify and invest in emerging companies that are developing transformative medicines and technologies that have the potential to enhance Pfizer’s pipeline and shape the future of our industry. For more information, visit www.pfizerventures.com.
About Bessemer Venture Partners
Bessemer Venture Partners is the world’s most experienced early-stage venture capital firm, with a portfolio of more than 200 companies, including Pinterest, Betterment, Smule, Rocket Lab, Procore, PagerDuty, Intercom, Fiverr, ServiceTitan, Toast, Allscripts, and Bright Health. Bessemer partners early with visionary entrepreneurs and supports them throughout every stage of their growth, primarily focusing on consumer, enterprise, healthcare, and frontier technology companies. The firm has backed more than 120 IPOs, including Shopify, Yelp, LinkedIn, Skype, OvaScience, LifeLock, Twilio, SendGrid, DocuSign, Wix, and MindBody. Bessemer’s 14 partners operate from offices in Silicon Valley, San Francisco, New York City, Boston, Israel, and India. For more information, visit www.bvp.com.
Contact:
Lissette Steele
Verge Scientific Communications
202.930.4762
lsteele@vergescientific.c
NEW YORK, Oct. 22, 2018 (GLOBE NEWSWIRE) -- Ovid Therapeutics Inc.(NASDAQ: OVID), a biopharmaceutical company committed to developing medicines that transform the lives of people with rare neurological diseases, today announced it has received the 2018 CDKL5 Forum Company Making a Difference Award for initiation of the Phase 2 ARCADE trial with OV935/TAK- 935, and its commitment to the CDKL5 deficiency disorder (CDD) patient community. The award was announced today in London, UK at the CDKL5 Forum, the largest annual conference on the advancement of science and therapeutic development for CDD.
CDD is a rare, severe, neurological disorder that causes frequent, treatmentresistent seizures in the first few months of life. CDD results in a constellation of severe challenges, including developmental delay and intellectual disability, movement disorder, difficulty sleeping, scoliosis, visual impairment, microcephaly and various gastrointestinal difficulties. There are currently no FDA-approved therapies for CDD.
“We hand out the Company Making a Difference Award annually to recognize industry partners for their commitment and support, and this year we are pleased to recognize the team at Ovid Therapeutics,” said Ana Mingorance, Ph.D., chief development officer of Loulou Foundation. “Since the day Ovid selected CDD as a condition to pursue, they have shown amazing support and care for our community, and we are excited to partner with such a humancentric organization.”
The ARCADE trial is a Phase 2 multi-center, open-label, pilot study designed to evaluate the treatment of OV935 in pediatric patients with epileptic seizures associated with CDD or Duplication 15q (Dup15q) syndrome. The first patients have already been enrolled into ARCADE. This study is part of a global collaboration with Takeda Pharmaceutical Company Limited.
“We are honored to be recognized for our dedication and efforts to bring a novel treatment option to the clinic for those with CDD and other rare epilepsies,” said Amit Rakhit, M.D., MBA, chief medical and portfolio management officer of Ovid Therapeutics. “It was important to everyone at Ovid that I travel to London to accept this award to show our unwavering support and partnership with the CDD community. We thank LouLou Foundation and the community and look forward to providing updates on our progress with the ARCADE study.”
The CDKL5 Forum is a unique community of collaboration and knowledge exchange, made up of leading scientists, clinicians and company representatives from around the world, united in the mission of better understanding the CDKL5 gene and disorder. The objective is to share current research on CDKL5 and to stimulate peer-group discussion and brainstorming around existing and future avenues of research and therapeutic approaches, in order to accelerate treatments and ultimately find cures for this neuro-genetic disorder. Now in its fourth year, the Forum represents the flagship annual event of the CDKL5 Program of Excellence, established by the Loulou Foundation and the Orphan Disease Center of the University of Pennsylvania’s Perelman School of Medicine.
About the ARCADE Trial
ARCADE is a Phase 2, multi-center, open-label, pilot study that will evaluate the treatment of OV935 in pediatric patients, aged 2 to 17 years old, with epileptic seizures associated with CDD or Dup15q syndrome. The primary endpoint is the change in motor seizure frequency in patients treated with OV935 by disorder (CDD and Dup15q). The key secondary endpoints include safety and tolerability, including percentage of participants considered treatment responders, change in CGI-S/C and correlation of OV935 concentration and plasma 24HC levels.
ARCADE is expected to enroll approximately 15 children with each condition at clinical trial sites in the United States. This study consists of a four to six week screening period to establish baseline seizure frequency followed by a 12-week treatment period (2-week dose titration and 10-week maintenance period.) To learn more about ARCADE visit clinicaltrials.gov or www.arcadestudy.com. At the end of treatment, eligible patients can roll over into the ENDYMION study. Additional details on the ENDYMION clinical trial can be found at www.clinicaltrials.gov.
About Cyclin-Dependent Kinase-Like 5 (CDKL5) Deficiency Disorder (CDD)
Cyclin-Dependent Kinase-Like 5 (CDKL5) deficiency disorder, also known as CDD, is a rare, severe, neurological disorder caused by mutations in the CDKL5 gene on the X-chromosome. The CDKL5 gene provides instructions for making a protein that is essential for normal brain and neuron development, and may play a role in regulating the activity of other genes. CDD causes early onset and treatment resistant epilepsy in the first few months of life. Other common features of CDD include severe developmental delay and intellectual disability, poor fine motor skills, difficulty sleeping, scoliosis, visual impairment, microcephaly and various gastrointestinal difficulties.
About Duplication 15q (Dup15q) Syndrome
Duplication 15q syndrome, also known as Dup15q syndrome, is a rare, severe, neurological disorder that results from duplications of chromosome 15q11.2-q13.1. In most cases, the chromosome mutation is not inherited but occurs during formation of reproductive cells or during embryonic development. Those with Dup15q syndrome experience seizures, hypotonia (poor muscle tone), developmental delays and intellectual disability. Difficult to control seizures are the most devastating symptom of Dup15qii. The severity of Dup15q and associated symptoms varies based on the size and location of the duplication and which genes are involved. There is insufficient demographic data to determine the prevalence of Dup15q in the general population.
About Investigational OV935/TAK-935
OV935/TAK-935 is a potent, highly-selective, first-in-class inhibitor of the enzyme cholesterol 24-hydroxylase (CH24H) being investigated as an anti-epileptic drug (AED). CH24H is predominantly expressed in the brain, where it plays a central role in cholesterol homeostasis. CH24H converts cholesterol to 24-hydroxycholesterol (24HC), which then exits the brain into the blood plasma circulation. Glutamate is one of the main neurotransmitters in the brain and has been shown to play a role in the initiation and spread of seizure activity. Recent literature indicates CH24H is involved in over-activation of the glutamatergic pathway through modulation of the NMDA channel, implying its potential role in central nervous system diseases such as epilepsy. Ovid and Takeda believe that OV935’s novel mechanism of action may potentially treat rare epilepsies by inhibiting CH24H to decrease 24HC levels, effectively decreasing glutamate hyperactivity. To Ovid and Takeda’s knowledge, OV935 is the only molecule with this mechanism of action in clinical development. OV935 is an investigational drug, not approved for commercial use.
OV935 has successfully completed four Phase 1 clinical studies, which have assessed tolerability, PK and target engagement at doses believed to be therapeutically relevant. In preclinical models, a novel proprietary PET ligand was used to determine target occupancy of OV935 in the brain. OV935 is being co-developed by Ovid and Takeda Pharmaceutical Company Limited.
The United States Food and Drug Administration (FDA) has granted orphan drug designation to OV935 for the treatment of both Dravet syndrome and LGS.
About Ovid Therapeutics
Ovid Therapeutics (NASDAQ: OVID) is a New York-based biopharmaceutical company using its BoldMedicine™ approach to develop medicines that transform the lives of patients with rare neurological disorders. Ovid has a broad pipeline of potential first-in-class medicines. The company’s lead investigational medicine, OV101, is currently in development for the treatment of Angelman syndrome and Fragile X syndrome. Ovid is also developing OV935/TAK-935 in collaboration with Takeda Pharmaceutical Company Limited for the treatment of rare developmental and epileptic encephalopathies (DEE).
For more information on Ovid, please visit http://www.ovidrx.com/.
Forward-Looking Statements
This press release includes certain disclosures that contain “forward-looking statements,” including, without limitation, statements regarding the potential clinical benefit of OV935 to treat patients with rare epilepsies, number of patients enrolled, the initiation, progress, timing, scope and results of clinical trials, and the effects of OV935 on efficacy, safety and tolerability. You can identify forward-looking statements because they contain words such as “will,” “believes” and “expects.” Forward-looking statements are based on Ovid’s current expectations and assumptions. Because forward-looking statements relate to the future, they are subject to inherent uncertainties, risks and changes in circumstances that may differ materially from those contemplated by the forward-looking statements, which are neither statements of historical fact nor guarantees or assurances of future performance. Important factors that could cause actual results to differ materially from those in the forward-looking statements are set forth in Ovid’s filings with the Securities and Exchange Commission under the caption “Risk Factors”. Ovid assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Contacts:
Patient advocacy:
Luke Rosen
Ovid Therapeutics Inc
lrosen@ovidrx.com
Media:
Jerica Pitts
W2O Group
jpitts@w2ogroup.com
(312) 858-3469
Ovid Therapeutics Inc.
New capital will enable Evidation Health to further enhance platform capabilities that quantify real life patient outcomes at scale
SAN MATEO, Calif.--(BUSINESS WIRE)--Today Evidation Health announced that it has closed a $10 Million funding round led by Sanofi-Genzyme BioVentures—the corporate venture capital arm of Sanofi, a global healthcare company that discovers, develops and distributes therapeutic solutions focused on patients' needs. Existing investors GE Ventures and B Capital Group also participated in the round.
The new investment will enable Evidation Health to further enhance its capabilities in large scale behavioral analytics, health outcomes measurement, and digital biomarker development using real life patient data from hundreds of applications and devices.
“At Evidation Health, we focus on measuring behavior-driven outcomes to help our partners understand and maximize product impact in the real world,” says Deborah Kilpatrick, CEO of Evidation Health. “We are excited about the Sanofi investment, as it aligns to our creation of faster, better ways to quantify patient outcomes—which we believe will directly fuel innovation across healthcare and life sciences.”
Evidation Health combines expertise from consumer technology, data science, outcomes research, and healthcare economics to measure product impact at entirely new levels of scale. By identifying what truly impacts patient outcomes outside of clinic walls, the value of all sorts of interventions can be quantified in the digital era of medicine.
“With the transition to value-based care, it is increasingly important for pharma companies to understand realworld behavior of individual patients and populations outside of the traditional clinical trial setting,” says Bernard Davitian, Vice President and Managing Director of Sanofi-Genzyme BioVentures. “Evidation Health’s unique capabilities in behavior analytics and integrated patient data are big enablers of this understanding. We’ve been impressed with both the Evidation team and platform, and we believe that they have the potential to transform the way pharma companies interact with patients to deliver better outcomes.”
About Evidation Health
Evidation Health helps healthcare companies quantify outcomes in the digital era, with real life data from connected patients. The company developed its Real Life Study Platform to accelerate and enhance outcomes research through virtualized pragmatic trials at scale—quantifying the impact of digital and traditional interventions far more efficiently than conventional approaches. Evidation Health works across the healthcare ecosystem with top pharmaceutical companies, payers, providers, and digital health companies. Evidation
Health is a privately held company headquartered in San Mateo, CA. For more information, visit www.evidation.com.
About Sanofi-Genzyme BioVentures
Sanofi-Genzyme BioVentures (SGBV) is the corporate venture capital arm of Sanofi. SGBV invests in early stage companies developing innovative products or technologies of interest to Sanofi. Today, SGBV has assembled a portfolio of direct equity investments in a variety of promising innovative life-science and digital health companies. SGBV is an important component of Sanofi's broader global strategy to invigorate external innovation. For more information, visit: http://sanofigenzymebioventures.com/

-Complete Responses Observed in Three of Six Patients with relapsed or refractory CD20-positive B cell non-Hodgkin Lymphoma at Dose Level 1 of the ATTCK-20-03 Phase I Study -
- No SAEs of Cytokine Release Syndrome or Neurotoxicity Observed in Dose Level 1 -
- Study Enrollment Continues -
-Management to host conference call today at 8:30 a.m. EDT-
CAMBRIDGE, Mass., Sept. 17, 2018 (GLOBE NEWSWIRE) -- Unum Therapeutics Inc. (NASDAQ: UMRX), a clinical-stage biopharmaceutical company focused on the development of cellular immunotherapies based on its novel, universal Antibody-Coupled T cell Receptor (ACTR) technology platform, today announced that the Company will be presenting data on the first dose level (40x106 ACTR+ T cells) of its ATTCK-20-03 clinical trial evaluating ACTR707 in combination with rituximab in patients with relapsed or refractory CD20-positive B cell non-Hodgkin Lymphoma (r/r NHL). Three of the six patients treated at the first dose level achieved a complete response, two of which remained ongoing at the time of the most recent data cut off. No dose-limiting toxicities (DLTs) were observed in any of the four DLT-evaluable patients, and no serious or severe adverse events of cytokine release syndrome or neurotoxicity were observed in any patients. These data will be presented at the Fourth Annual CRI-CIMT-EATI-AACR International Cancer Immunotherapy Conference on September 30 in New York, New York.
“These preliminary clinical data suggest that complete responses may be achieved without cytokine release syndrome, further validating the potential of our proprietary ACTR technology platform,” said Chuck Wilson, Chief Executive Officer of Unum. “The ATTCK-20-03 trial is a key element of our strategy to develop novel therapeutics for patients with no available treatment options.”
“Complete responses observed in this heavily pre-treated patient population at the first dose level of the ATTCK-20-03 trial support the potential potency of ACTR T cells for these patients,” said Michael Vasconcelles, Chief Medical Officer of Unum. “Based upon this encouraging preliminary profile, we look forward to continuing the dose escalation phase of this study. We expect these and future data to support selection of an ACTR product candidate to progress into potential registration trials in patients with r/r NHL.”
Enrollment and ACTR707 dosing in the second dose cohort (60x106 ACTR+ T cells) of the ATTCK-20-03 trial has been completed and dose escalation is proceeding. The Company expects to present additional data from this study later this year.
Safety and Preliminary Efficacy of ACTR707, Autologous T Lymphocytes Expressing an Antibody-Coupled T Cell Receptor, in Combination with Rituximab in Subjects with Relapsed or Refractory CD20-Positive B-cell Lymphoma (Abstract #A003)
Presenter: Dr. Veronika Bachanova, University of Minnesota
Date: Sunday, September 30, 2018, 11:45am – 2:15pm
Location: New York Marriott Marquis, Westside Ballroom
ATTCK-20-03 is a Phase I, multi-center, open label, single arm clinical trial evaluating ACTR707 in combination with rituximab in patients with r/r NHL. Eligible patients for enrollment must have, among other criteria, received adequate prior anti-lymphoma therapy, including rituximab and chemotherapy, for their CD20-positive r/r NHL. Key eligibility criteria include: pre-specified eligible NHL subtypes, including DLBCL, disease progression following immediate prior therapy, adequate organ function and performance status, and measurable disease. The trial design includes a dose escalation phase using an adaptive design, followed by a cohort expansion phase. Primary study objectives are to characterize the safety of ACTR707 in combination with rituximab and to determine the maximum tolerated dose and proposed recommended Phase 2 dose. Secondary study objectives include: assessment of the anti-lymphoma activity of the combination, ACTR707 persistence, rituximab pharmacokinetics, and inflammatory markers and cytokine levels. Following leukapheresis, each patient receives lymphodepletion followed by the first infusion of rituximab and then a single infusion of ACTR707. Rituximab infusions continue on a regular, pre-specified schedule.
Data in the first cohort of the trial demonstrate complete responses at the first response assessment in 3/6 patients treated with ACTR707 in combination with rituximab, two of which remained ongoing at the time of the most recent data cut off. There were no serious or severe adverse events of cytokine release syndrome, neurotoxicity, or autoimmune events. Grade 3 or higher adverse events were mostly hematologic including neutropenia (n=2), febrile neutropenia (n=2), and thrombocytopenia (n=1). ACTR+ T cells were detectable in all subjects and ACTR+ T cells persisted in the presence of continued rituximab administration. These results support the continued dose escalation of ACTR707 in combination with rituximab.
Conference Call and Webcast
Unum will host a conference call and webcast at 8:30 a.m. EDT today to discuss the data. To participate in the conference call, please dial (866) 300-3411 (domestic) or (636) 812-6658 (international) and enter the conference code: 2958105. To join the live webcast, please visit the investor relations section of the Unum Therapeutics website at https://investors.unumrx.com/ at least 10 minutes before the event begins. A webcast replay will be available at the same location on the Unum Therapeutics website beginning approximately two hours after the event and will be archived for 90 days.
About ACTR707
ACTR707 is an investigational drug that may represent an important construct not only for adult patients with CD20+ r/r NHL, when used in combination with rituximab, but also for patients with other cancer types when used in combination with other antibodies. ACTR707 was identified through a comprehensive high-throughput screening effort aimed at identifying receptors with improved functional characteristics across several dimensions. In preclinical testing, ACTR707 demonstrated potent activity against a wide range of hematologic and solid tumor cancers. Given the challenges of the immunosuppressive solid tumor microenvironment, Unum believes that ACTR707’s increased activity may be particularly important in addressing solid tumor cancers. ACTR707 is currently being tested in combination with rituximab in patients with r/r NHL in a Phase I multi-center open label clinical trial, ATTCK-20-03. Testing is expected to be initiated later in 2018 in ATTCK-34-01, a Phase I multi-center open label clinical trial exploring the combination of ACTR707 with trastuzumab in patients with HER2+ advanced cancers.
About Unum Therapeutics
Unum Therapeutics is a clinical-stage biopharmaceutical company focused on the development and commercialization of novel immunotherapy products designed to harness the power of a patient’s immune system to cure cancer. Unum’s novel proprietary technology, antibody-coupled T cell receptor (ACTR) is a universal, engineered cell therapy intended to be used in combination with a wide range of tumor-specific antibodies to target different tumor types. ACTR087 and ACTR707, each used in combination with rituximab, an anti-CD20 antibody, are Unum’s two most advanced product candidates, currently in Phase I clinical testing in adult patients with relapsed or refractory non-Hodgkin lymphoma (r/r NHL). The Company has an additional product candidate in Phase I clinical testing: ACTR087 used in combination with the novel antibody SEA-BCMA in adult patients with relapsed or refractory multiple myeloma. Finally, the Company has an active investigational new drug application (IND) for ACTR707 used in combination with trastuzumab, an anti-human epidermal growth factor receptor 2 (HER2) antibody, to treat patients with HER2+ advanced cancer. This Phase I trial is expected to be initiated by the end of 2018.
The Company is headquartered in Cambridge, MA.
Forward looking Statements
This press release contains forward-looking statements. Statements in this press release about our future expectations, plans and prospects, our long-term growth, the anticipated timing of our clinical trials and regulatory filings, the development of our product candidates, including the four lead ACTR product candidates, as well as other statements containing the words "anticipate," "believe," "continue," "could," "estimate," "expect," "intend," "may," "might," "plan," "potential," "predict," "project," "should," "target," "will," or "would" and similar expressions, constitute forward-looking statements within the meaning of the safe harbor provisions of The Private Securities Litigation Reform Act of 1995. We may not actually achieve the forecasts disclosed in our forward-looking statements, and you should not place undue reliance on our forward-looking statements. Actual results could differ materially from the projections disclosed in the forward-looking statements we make as a result of a variety of risks and uncertainties, including risks related to the accuracy of our estimates regarding expenses, future revenues, capital requirements, and the need for additional financing, the success, cost and timing of our product development activities and clinical trials, our ability to obtain and maintain regulatory approval for our product candidates, and the other risks and uncertainties described in the "Risk Factors" sections of our public filings with the Securities and Exchange Commission. In addition, the forward-looking statements included in this press release represent our views as of the date hereof. We anticipate that subsequent events and developments may cause our views to change. However, while we may elect to update these forward-looking statements at some point in the future, we specifically disclaim any obligation to do so. These forward-looking statements should not be relied upon as representing our views as of any date subsequent to the date hereof.
Investor Contact:
Stephanie Ascher, 212-362-1200
stephanie@sternir.com
Media Contact:
Paul Kidwell, 617-680-1088
paul.kidwell@unumrx.com
Proceeds will help advance lead program in facioscapulohumeral muscular dystrophy (FSHD) and further support advancement of rare disease-focused pipeline
Cambridge, Mass., September 5, 2018 – Fulcrum Therapeutics, a company focused on discovering and developing small molecule therapies to unlock gene control and treat serious genetic diseases, today announced the closing of an $80 million Series B financing. Proceeds from the financing will be used to advance Fulcrum’s lead program in facioscapulohumeral muscular dystrophy (FSHD) into clinical testing, and to progress its pipeline of therapeutics for rare, genetically based neuromuscular, central nervous system and hematologic disorders.
The financing was led by Foresite Capital, with participation by [Fidelity Management and Research Company, 6 Dimensions Capital, Casdin Capital, Sanofi Ventures, Section 32, NS Investments, entities affiliated with Leerink Partners, and undisclosed institutional investors. Jim Tananbaum, M.D., of Foresite Capital will join the Board of Directors of Fulcrum.
“We are delighted to have the support of this exceptional group of investors as we continue to work towards our vision of bringing new futures to patients and families affected by debilitating genetic diseases,” said Robert J. Gould, Ph.D., Fulcrum’s President and Chief Executive Officer. “This funding will provide crucial support as we move rapidly towards the clinic with our lead drug candidate in FSHD, and further advance our pipeline of small molecule therapies.”
Fulcrum is pioneering a small molecule, precision medicine approach to address severe monogenic and prevalent diseases of gene misregulation. By focusing on disease-causative genes, the company is unlocking the druggable mechanisms that regulate disease to develop a new generation of therapies in multiple therapeutic areas. In partnership with the FSHD Clinical Trial Research Network (CTRN), the company has initiated a clinical trial readiness study in FSHD. The aim of the study is to standardize a set of tools and measurements for Fulcrum’s future clinical drug trials.
“Fulcrum’s uniquely patient-centered approach to treating severe genetic diseases is already delivering strong early results,” said Dr. Tananbaum. “We are confident that Fulcrum will continue to lead the way in developing novel medicines for patients and families who have no viable treatment options, and we look forward to supporting the team as it works to further advance its rare-disease focused pipeline.”
About Fulcrum Therapeutics
Fulcrum Therapeutics is a biotechnology company developing new medicines to deliver a new future to patients and their families by transforming gene regulation in disease. Fulcrum’s therapies are based on modulating gene regulation via control of genetic on and off switches of disease genes. Fulcrum, headquartered in Cambridge, Mass., was launched by Third Rock Ventures in 2016 and named a “Fierce 15” company later that year. For more information, please visit www.fulcrumtx.com.
CONTACTS:
Media: Sarah Sutton
Ten Bridge Communications
sarah@tenbridgecommunications.com
518-932-3680
T1D Sleep Pilot to study the relationship between low blood sugar at night and daily behavior, as measured by smartphones and connected sensors from wearables and medical devices
SAN MATEO, CALIF., August 23, 2018 – Evidation Health, a health and measurement company that helps life sciences and health care companies understand how everyday behaviors and health interact, announced today a new technology partnership with Tidepool, an open source, not-for-profit company focused on making diabetes data more accessible, actionable and meaningful for people with diabetes, their care teams and researchers.
Under the partnership, Evidation and Tidepool are joining together on a new research study, called the T1D Sleep Pilot, to capture and study real world data from people with type 1 diabetes. The research will develop insights from data generated by continuous glucose monitors and insulin pumps, in addition to sleep and activity trackers from smartphones and other connected sensors.
The observational study will explore linkages between nocturnal hypoglycemia, next-day behavior, sleep patterns, and heart rates in order to contribute to ongoing research on how everyday behavior data and diabetes complications interact.
“Linking real world data from connected devices with other medical data in virtual studies allow us to measure how behaviors — outside of the doctor’s office or hospital — affect health and impact outcomes,” said Evidation Health CEO Deborah Kilpatrick, Ph.D. “Tidepool has done a fantastic job of giving people with diabetes access to their own data, and this effort provides a new opportunity to share everyday data with researchers at scale and enable more people to contribute to diabetes innovation.”
The partnership brings together Evidation’s data platform, which analyzes and processes large-scale sensor and behavior data in clinically meaningful ways, with Tidepool’s device-agnostic consumer software. Tidepool joins a growing list of more than 100 individually-permissioned data sources that are linked to Evidation’s platform, including Apple Health, Blue Button, Dexcom, Epic and Fitbit.
“People with diabetes can use their individual data to play a key role in improving health,” said Howard Look, president, CEO, and founder of Tidepool. “Our study with Evidation gives people with diabetes a new way to share their data with researchers, and contribute to a better understanding of dangerous low-blood sugar levels, which can often occur more frequently while sleeping.”
Since 2012, Evidation Health has built the most diverse virtual pool of research participants through its proprietary app, Achievement. With more than 2 million individuals using the app, this cohort is unparalleled in clinical research today.
People with type 1 diabetes can learn more about joining the T1D Sleep Pilot at http://study.myachievement.com/t1dsleep.
About Evidation
Evidation Health is a new kind of health and measurement company that provides the world’s most innovative life sciences and health care companies the technology and expertise they need to understand how everyday behavior and health interact. The volume of behavior data generated from smartphones and connected sensors — including wearables and medical devices — has opened up new ways to analyze individuals’ behavior and health in real time, unlocking insights into what medicines and treatments work best and spotting significant changes in health earlier. The scale and utility of everyday behavior data has the potential to be one of the most transformative forces in medicine, and Evidation Health is leading the way. Over the years, Evidation has built the largest, most diverse virtual pool of research participants through its proprietary and popular app, Achievement. With a direct and trusted relationship with more than 2 million individuals, its deep research expertise, and its data platform, Evidation Health can undertake real world research for life sciences and health care companies — and, ultimately, transform how health is measured and how diseases are identified, treated, and monitored.
Founded in 2012, Evidation Health is headquartered in San Mateo, Calif., with additional offices in San Francisco and Santa Barbara, Calif. To learn more, visit evidation.com, or follow us on Twitter @evidation.
About Tidepool
Tidepool is an open source, not-for-profit company focused on liberating data from diabetes devices, supporting researchers, and providing great, free software to people with diabetes and their care teams. Through the Tidepool Clinical Studies Platform and the Tidepool Big Data Donation Project, Tidepool is empowering the next generation of diabetes research and innovation. To learn more, visit tidepool.org.
Share on LinkedIn Twitter
---- Deborah Kilpatrick, Ph.D.
CEO
Evidation Health | www.evidation.com
650.906.7641
- Poster presentation of STARS topline data: the first clinical trial to show a positive clinical
benefit on overall Angelman syndrome impairments -
NEW YORK, Aug. 16, 2018 (GLOBE NEWSWIRE) -- Ovid Therapeutics Inc. (NASDAQ: OVID), a biopharmaceutical company committed to developing medicines that transform the lives of people with rare neurological diseases, today announced that an abstract on OV101 was accepted for poster presentation at the 65th American Academy of Child and Adolescent Psychiatry (AACAP) Annual Meeting taking place in Seattle, Wash., October 22 to 27, 2018. AACAP is the largest international gathering of child and adolescent psychiatrists.
Angelman syndrome is a rare, lifelong, genetic disorder that affects 1 in 15,000 people in the general population. It is characterized by severe impairment in behavior, learning, verbal communication, motor skills, and sleep, and there are no FDA-approved medicines or an established treatment paradigm for this condition. If approved, OV101 could be the first medicine to specifically target a key underlying neurological dysfunction of Angelman syndrome: impaired tonic inhibition, which is most commonly caused by a disruption of the ubiquitin protein ligase (UBE3A) gene.
Poster presentation details
Title: Topline Results from a Phase 2 Adult and Adolescent Angelman Syndrome Clinical Trial: A Randomized, Double-Blind, Safety and Efficacy Study of Gaboxadol (OV101)
Session: New Research Poster Session 3
Date and Time: Thursday, October 25, 2018, 9:30 a.m. – 12:00 p.m. ET
Topline data from the STARS trial were announced on August 6, 2018.
About OV101
OV101 (gaboxadol) is believed to be the only delta (δ)-selective GABAA receptor agonist in development and the first investigational drug to specifically target the disruption of tonic inhibition, a central physiological process of the brain that is thought to be the underlying cause of certain neurodevelopmental disorders. OV101 has been demonstrated in laboratory studies and animal models to selectively activate the δ-subunit of GABAA receptors, which are found in the extrasynaptic space (outside of the synapse), and thereby impact neuronal activity through tonic inhibition.
Ovid is developing OV101 for the treatment of Angelman syndrome and Fragile X syndrome to potentially restore tonic inhibition and relieve several of the symptoms of these disorders. In preclinical studies, it was observed that OV101 improved symptoms of Angelman syndrome and Fragile X syndrome. This compound has also previously been tested in over 4,000 patients (over 1,000 patient-years of exposure) and was observed to have favorable safety and bioavailability profiles.
The FDA has granted Orphan Drug and Fast Track designations for OV101 for both the treatment of Angelman syndrome and Fragile X syndrome. The U.S. Patent and Trademark Office has granted Ovid patents directed to methods of treating Angelman syndrome and Fragile X syndrome using OV101. The issued patents expire in 2035.
About Ovid Therapeutics
Ovid Therapeutics (NASDAQ: OVID) is a New York-based biopharmaceutical company using its BoldMedicine™ approach to develop medicines that transform the lives of patients with rare neurological disorders. Ovid has a broad pipeline of first-in-class medicines. The company’s lead investigational medicine, OV101, is currently in development for the treatment of Angelman syndrome and Fragile X syndrome. Ovid is also developing OV935/TAK-935 in collaboration with Takeda Pharmaceutical Company Limited for the treatment of rare developmental and epileptic encephalopathies (DEE).
For more information on Ovid, please visit http://www.ovidrx.com/.
Forward-Looking Statements
This press release includes certain disclosures that contain “forward-looking statements,” including, without limitation, statements regarding (i) the potential clinical benefit of OV101 to treat patients with Angelman syndrome, and (ii) the timing and results of any discussions with regulatory authorities regarding the registrational path for OV101 and approval. You can identify forward-looking statements because they contain words such as “will,” “believes” and “expects.” Forward-looking statements are based on Ovid’s current expectations and assumptions. Because forward-looking statements relate to the future, they are subject to inherent uncertainties, risks and changes in circumstances that may differ materially from those contemplated by the forward-looking statements, which are neither statements of historical fact nor guarantees or assurances of future performance. Important factors that could cause actual results to differ materially from those in the forward-looking statements are set forth in Ovid’s filings with the Securities and Exchange Commission. Ovid assumes no obligation to update any forwardlooking statements contained herein to reflect any change in expectations, even as new information becomes available.
Contacts
Investors:
Lora Pike
Ovid Therapeutics Inc.
Senior Director, Investor Relations & Public Relations
lpike@ovidrx.com
Steve Klass
Burns McClellan, Inc
sklass@burnsmc.com
(212) 213-0006
Media:
Elliot Fox
W2O Group
efox@w2ogroup.com
(212) 257-6724
– First Solid Tumor Product Candidate Based on Unum’s universal ACTR Technology –
– Phase 1 Study Expected to Initiate by the End of 2018 –
CAMBRIDGE, Mass., Aug. 13, 2018 (GLOBE NEWSWIRE) -- Unum Therapeutics Inc. (NASDAQ: UMRX), a clinical-stage biopharmaceutical company focused on the development of cellular immunotherapies based on its novel, universal Antibody-Coupled T Cell Receptor (ACTR) technology platform, today announced that an investigational new drug (IND) application is now active for ACTR T cells in combination with trastuzumab for the treatment of patients with HER2+ advanced cancers. This represents the first solid tumor product candidate
based on Unum’s novel, universal ACTR technology, and the fourth clinical trial program for the Company.
“We are very happy to reach this important milestone for patients and for Unum,” said Chuck Wilson, Chief Executive Officer of Unum. “ACTR represents a promising novel technology that can be used to target different tumor types and it’s exciting to expand its application to target solid tumors. We are committed to developing ACTR for patients with HER2+ advanced cancers who need better treatment options.”
Under this IND, Unum is preparing to initiate a multi-center Phase I trial, called ATTCK-34-01, by the end of 2018 in patients with HER2+ advanced cancers. ATTCK-34-01 is designed as a dose escalation study where both the ACTR T cell drug product and trastuzumab doses are escalated in order to define the safety, tolerability, and anti-tumor activity of the combination. Expansion at the recommended Phase 2 dose is planned.
About Unum Therapeutics
Unum Therapeutics is a clinical-stage biopharmaceutical company focused on the development and commercialization of novel immunotherapy products designed to harness the power of a patient’s immune system to cure cancer. Unum’s novel proprietary technology, antibody-coupled T cell receptor (ACTR), is a universal, engineered cell therapy intended to be used in combination with a wide range of tumor-specific antibodies to target different tumor types. ACTR087 used in combination with rituximab, an anti-CD20 antibody, is Unum’s most advanced product candidate, currently in Phase I clinical testing in adult patients with relapsed or refractory non-Hodgkin lymphoma (r/r NHL). The Company has two additional product candidates in Phase I clinical testing: ACTR087 used in combination with the novel antibody SEA-BCMA in adult patients with relapsed or refractory multiple myeloma and ACTR707, a modified ACTR construct, used in combination with rituximab in adult patients with r/r NHL. Finally, the Company has an active investigational new drug application (IND) for ACTR707 used in combination with trastuzumab, an anti- human epidermal growth factor receptor 2 (HER2) antibody, to treat patients with HER2+ cancers and expects to initiate the Phase 1 trial by the end of 2018.
The Company is headquartered in Cambridge, MA.
Forward looking Statements
This press release contains forward-looking statements. Statements in this press release about the Company’s future expectations, plans and prospects, including projections regarding the anticipated timing of its clinical trials and regulatory filings, the development of its product candidates, including the four lead ACTR product candidates, as well as other statements containing the words "anticipate," "believe," "continue," "could," "estimate," "expect," "intend," "may," "might," "plan," "potential," "predict," "project," "should," "target," "will," or "would" and similar expressions, constitute forward-looking statements within the meaning of the safe harbor provisions of The Private Securities Litigation Reform Act of 1995. The Company may not actually achieve the forecasts disclosed in the Company’s forward-looking statements, and undue reliance should not be placed on its forward-looking statements. Actual results could differ materially from the projections disclosed in the forward-looking statements the Company makes as a result of a variety of risks and uncertainties, including risks related to the accuracy of its estimates regarding expenses, future revenues, capital requirements, and the need for additional financing, the success, cost and timing of its product development activities and clinical trials, its ability to obtain and maintain regulatory approval for its product candidates, and the other risks and uncertainties described in the "Risk Factors" sections of the Company’s public filings with the Securities and Exchange Commission. In addition, the forward-looking statements included in this press release represent the Company’s views as of the date hereof. The Company anticipates that subsequent events and developments may cause its views to change. However, while it may elect to update these forward-looking statements at some point in the future, it specifically disclaims any obligation to do so. These forward-looking statements should not be relied upon as representing the Company’s views as of any date subsequent to the date hereof.
Investor Contact:
Stern Investor Relations, Inc.
Mary T. Conway, 212-362-1200
mary@sternir.com
Media Contact:
Paul Kidwell, 617-680-1088
paul.kidwell@unumrx.com
Former CEO and President of Zafgen brings pharmaceutical development and public biotechnology company
leadership experience as Navitor builds product pipeline
George P. Vlasuk, Ph.D. to remain as President and appointed Chief Scientific Officer
CAMBRIDGE, Mass., August 2, 2018 – Navitor Pharmaceuticals, Inc., a biopharmaceutical company targeting the mTORC1 pathway to develop novel therapeutics that help patients live longer and healthier lives, today announced the appointment of Thomas E. Hughes, Ph.D., as Chief Executive Officer. Dr. Hughes is the former Chief Executive Officer of Zafgen (Nasdaq:ZFGN), a publicly‐traded biotechnology company developing medicines for metabolic diseases and has been a scientific advisor to Navitor since its seed stage. George P. Vlasuk, Ph.D., current President and CEO of Navitor, will remain President and will assume the newly‐created role of Chief Scientific Officer. Both Dr. Hughes and Dr. Vlasuk will serve on Navitor’s Board of Directors.
These moves come at an exciting time for Navitor, which recently initiated a Phase 1 clinical study of its lead candidate, NV‐5138, for treatment‐resistant depression (TRD) and is advancing its mTORC1 platform to address multiple therapeutic applications, including CNS, immuno‐metabolism, fibrosis and multiple rare diseases.
“We are delighted to bolster the leadership team at Navitor to further strengthen our ability to advance a multi‐product pipeline based on our proprietary mTORC1 platform. Tom brings extensive pharmaceutical development and public biotechnology company experience to lead this next stage of Navitor’s growth,” said Alan Crane, Co‐founder and Chairman of the Board of Navitor and Entrepreneur Partner at Polaris Partners.
“George has been instrumental in building Navitor to establish the industry‐leading capabilities of our mTORC1 platform. George had the foresight to propose bringing a CEO on board with public company and translational experience, while he looks to focus his efforts on continuing to advance the science of Navitor. We are thrilled to have both George and Tom as members of the Navitor leadership team and are confident that with their combined experience, we will be able to advance the company to its next stage of evolution,” continued Mr. Crane.
“I am excited to join Navitor and am eager to work with George and the team he has built to move Navitor through its next stages of growth,” said Dr. Hughes. “mTORC1 is a powerful biological node with proven and tractable approaches to drug development. With a strong platform of proprietary approaches to drugging the mTORC1 system both for selective inhibitors and activators, Navitor is well positioned to lead this field of biology with valuable new insights and therapeutic programs addressing diseases with high and unmet medical need. The recent clinical trial initiation of NV‐5138 is the first application of Navitor’s mTORC1 technology in its platform of programs that aim to bring new therapies to patients with limited treatment options.”
Navitor’s small molecule therapeutics are designed to selectively modulate the cellular signals that are aberrant in disease processes caused by the dysregulation of mTORC1 activation. Navitor was founded based on the groundbreaking discoveries related to the mTORC1 pathway and nutrient signaling mechanisms by Dr. David Sabatini at The Whitehead Institute for Biomedical Research.
Dr. Vlasuk commented, “I am excited to focus my efforts on further building the capabilities of Navitor’s mTORC1 platform, expanding our pipeline and shaping the long‐term direction of our drug discovery and development efforts. I look forward to working with Tom, whom I have known professionally for many years, to realize our Company’s vision.”
Dr. Tom Hughes, who has more than 30 years of industry experience in the development and commercialization of pharmaceutical products, most recently served as President and Chief Scientific Officer of Zafgen and previously led Zafgen as CEO from 2008 to 2017. During this time, Dr. Hughes established Zafgen as a leading biotechnology company working in the area of rare and prevalent metabolic disorders and led the company through its Initial Public Offering in 2014. Prior to Zafgen, Dr. Hughes held several positions at Novartis including Global Head of the Cardiovascular and Metabolic Diseases Therapeutic Area at the Novartis Institutes for BioMedical Research in Cambridge, MA. In these roles, he oversaw many drug discovery and development projects targeting major global aging‐related health issues including obesity, diabetes, and heart disease. Dr. Hughes currently serves as a member of the Board of Directors of miRagen Therapeutics, Inc., is an advisor to Atlas Venture, and is a member of several scientific and strategic advisory boards, including Broadview Ventures, HotSpot Therapeutics, and Nimbus Therapeutics. He holds a Ph.D. in nutritional biochemistry from Tufts University, an M.S. in zoology from Virginia Polytechnic Institute & State University and a B.A. in biology from Franklin and Marshall College.
About NV‐5138
NV‐5138 is an orally bioavailable, small molecule that is designed to directly and transiently activate mTORC1 activity by binding to and modulating a newly discovered cellular sensor protein for the amino acid leucine, which is a potent natural activator of mTORC1. Unlike leucine, oral administration of NV‐5138 results in significant mTORC1 pathway activation in the brain since it is not broken down or incorporated into new proteins. These properties make NV‐5138 a unique agent with which to evaluate the role of mTORC1 in brain disorders, such as depression, where mTORC1 activity is often suppressed. Results from preclinical models demonstrate that NV‐5138 produces rapid upregulation of key synaptic proteins, synaptogenesis and sustained antidepressant behavioral responses via the transient and direct activation of the mTORC1 signaling pathway. Since NV‐5138 does not directly modulate the NMDA receptor pathway, it may not have the side effects and abuse potential observed with several NMDA receptor therapeutics currently in development. NV‐ 5138 is currently being clinically studied for the treatment of major depressive disorder (MDD) with an initial focus on treatment‐resistant depression (TRD).
About Navitor
Navitor Pharmaceuticals, Inc.is realizing the potential of modulating mTORC1, the master regulator of cellular function, to develop a pipeline of therapeutics that help patients live longer and healthier lives. Our industry leading team is unlocking the promise of recent discoveries in mTORC1 biology to address a broad range of chronic diseases. Our initial clinical application is a first‐in‐class drug to address unmet needs in depression. For more information, please visit www.navitorpharma.com.
Contact:
The Yates Network
Kathryn Morris, 914‐204‐6412
kathryn@theyatesnetwork.com
Launches First, Purpose-Built Data Platform to Measure and Better Understand How Everyday Behavior Data, Medical Conditions, and Symptoms Interact
SAN MATEO, CALIF., August 1, 2018 – Evidation Health, a health and measurement company that helps life sciences and health care companies understand how everyday behaviors and health interact, announced today that it has raised $30 million in Series C funding.
The round was co-led by SV Health Investors, a new investor, and existing investor B Capital Group. It included participation from existing investors GE Ventures and Sanofi Ventures. With this funding, Evidation has raised a total of $61 million.
Evidation also announced that it launched a new data platform to enable its customers to analyze and process large-scale sensor and behavior data in clinically meaningful ways.
“Our new data platform makes it easier for statisticians and data scientists at life sciences and health care companies to take everyday behavior and health care data, analyze it, and create a new understanding of health,” said Evidation Health CEO Deborah Kilpatrick, Ph.D. “This will help Evidation speed a transformation in real world research and knowledge, so we can better treat, diagnose, and predict the onset of disease.”
By linking real world data from smartphones and connected sensors — including wearables and medical devices — with traditional medical data, Evidation measures how behaviors outside of the doctor’s office or hospital relate to health and impact outcomes. The new data platform — which has been battle tested in a pilot program with a leading, global pharmaceutical company — can ingest individually-permissioned data from more than 100 sources ranging from Apple Health to Fitbit, Epic, Blue Button, and Dexcom.
The platform marries this information with traditional data from insurance claims, electronic health records, and diagnostic reports, alongside patient-reported outcomes collected directly through Evidation’s platform. This provides a new opportunity to analyze individual behavior and health in real time, not just when someone checks in with a physician or stops to record what they did or felt.
“Evidation’s data platform is opening up exciting new possibilities in medical research and the way we treat and prevent disease,” said P. Murali Doraiswamy, M.D., professor of psychiatry and medicine at Duke University Health System, and an advisor to Evidation. “With its unique analytics and methods, Evidation is transforming data into the kind of findings that traditional clinical trials may need substantially longer time frames, maybe even decades, to achieve. This will help identify health risks earlier and better treat the kinds of chronic diseases that scientists are working to cure.”
The secure, private platform currently processes more than 1 trillion data points each year across millions of individuals. It is built to manage the scale of high volume, continuous data streams and eliminates the need for infrastructure, data, and feature engineering on the part of Evidation customers, allowing them to focus entirely on analytics.
Already, the platform is being used to invent new ways to measure health across diabetes and cognitive decline. This complements the work that the company is performing with partners across multiple therapeutic areas ranging from cardiovascular disease to chronic pain, rheumatoid arthritis, migraine, depression and anxiety, fatigue, heart failure, and asthma — to name a few. In addition to supporting the new data platform, the funding will be used to develop partnerships, adding to Evidation’s vast array of real world data sources, and to launch novel research studies to better understand and measure how everyday behavior and health interact.
“We’re excited to deepen our partnership with Evidation Health and continue supporting the team as they build the leading platform to generate clinical relevance from everyday behavior data in real world populations. Evidation’s ability to link patient behavior to medical outcomes is truly revolutionary and will help transform how we measure health in everyday life,” said Raj Ganguly, co-founder and partner of B Capital Group.
“Evidation stands out not only because of its powerful data intake, aggregation, and analytics capabilities, but also because of the strength and vision of the company’s leadership team with a rare combination of deep technology and health care expertise. Evidation represents a unique platform company with the ability to meaningfully improve clinical development and, by extension, drug delivery, treatment, and outcomes,” added SV Health Investors Managing Partner Michael Balmuth, who is joining Evidation’s board of directors.
Since 2012, Evidation Health has built the most diverse virtual pool of research participants through its proprietary app, Achievement. With more than 2 million individuals using the app, this cohort is unparalleled in clinical research today, and represents the U.S.’s largest virtual research site. Individuals have the opportunity to participate in research studies, enabling Evidation to drill down to the individual level to address new questions and generate new context as needed — to measure what treatments work best and under what conditions, and enable earlier diagnoses and better matched interventions.
About Evidation
Evidation Health is a new kind of health and measurement company that provides the world’s most innovative life sciences and health care companies the technology and expertise they need to understand how everyday behavior and health interact. The volume of behavior data generated from smartphones and connected sensors — including wearables and medical devices — has opened up new ways to analyze individuals’ behavior and health in real time, unlocking insights into what medicines and treatments work best and spotting significant changes in health earlier. The scale and utility of everyday behavior data has the potential to be one of the most transformative forces in medicine, and Evidation Health is leading the way. Over the years, Evidation has built the largest, most diverse virtual pool of research participants through its proprietary and popular app, Achievement. With a direct and trusted relationship with more than 2 million individuals, its deep research expertise, and its data platform, Evidation Health can undertake real world research for life sciences and health care companies — and, ultimately, transform how health is measured and how diseases are identified, treated, and monitored. Founded in 2012, Evidation Health is headquartered in San Mateo, Calif., with additional offices in San Francisco and Santa Barbara, Calif. To learn more, visit evidation.com, or follow us on Twitter @evidation.
About SV Health Investors
V Health Investors, formerly named SV Life Sciences, is a healthcare and life sciences venture capital and growth equity firm. SV targets early-stage opportunities in biotechnology; early-stage and revenue-stage opportunities in medical devices; and growth equity investments for later-stage businesses in healthcare services and digital health. Over the past 20 years, SV Health Investors has invested in more than 175 companies. The firm currently has over $2.5 billion of capital commitments under management. SV Health Investors has offices in Boston and London. For more information, please visit www.svhealthinvestors.com.
About B Capital Group
B Capital Group is a global venture capital firm that invests in pioneering healthcare, fintech, industrial logistics and consumer enablement companies that are primed to scale across the global stage. Founded in partnership with The Boston Consulting Group, B Capital Group delivers unique access to top corporations to match cutting-edge start-ups with the world's leading CEOs, platforms, and brands. Existing portfolio companies include AImotive, Atomwise, Bird, Bizongo, Bright.md, Capital Match, Carro, CXA, Evidation Health, Fishbrain, Hollar, Icertis, INTURN, Journera, Lanetix, Mswipe, Myia, Ninja Van and SilverCloud Health. For more information, visit www.bcapgroup.com.
CAMBRIDGE, Mass. and NEW YORK - July 23, 2018 - Click Therapeutics, Inc. (“Click”), a leader in Digital Therapeutics™ solutions as prescription medical treatments, announces a $17 million financing round led by Sanofi Ventures. Click will use this financing to continue advancing its proprietary platform and pipeline of prescription digital therapeutics to treat a wide range of diseases. Recent notes were converted to equity as part of the financing.
“We’ve evaluated many companies in this space, and we believe Click Therapeutics’ mobile patient engagement platform positions the company to be a leader in the field of prescription digital therapeutics,” said Bernard Davitian, SVP and Managing Director of Sanofi Ventures, who has joined Click’s Board in conjunction with the financing. “Click’s platform enables the company to target multiple indications efficiently and effectively, and we have invested with the intent of partnering across a variety of therapeutic areas. Sanofi Ventures is excited to join Click on this journey of building a new pillar of medicine.”
In addition to Clickotine®, Click’s commercial product for smoking cessation, the company is developing prescription digital therapeutics for the treatment of depression (CT-152), insomnia (CT-141), acute coronary syndrome (CT-111), and chronic pain (CT-130). Click will seek FDA clearance for these programs as class II medical devices with disease-specific treatment claims, to be prescribed by physicians and reimbursed by payers.
“The Click Therapeutics team is proud to partner with Sanofi Ventures to advance our pipeline and expand our product portfolio of prescription medical treatments,” remarked David Benshoof Klein, Co-founder and CEO of Click. “As we announced last summer, in 2017 we expanded our collaboration with Magellan Health, Inc. to pursue regulatory clearance from the FDA for indication-specific prescription digital therapies, leveraging the industry-leading suite of intellectual property and data from Magellan’s existing software as well as their vast coverage and reimbursement leadership. The addition of Sanofi as a strategic investor, and the closing of this financing, represent major steps forward for Click and for the field of software as prescription medical treatments. By connecting patients with cognitive and neurobehavioral interventions, our platform will bring clinically-validated digital therapeutic solutions into mainstream healthcare.”
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi. Sanofi Ventures invests in early-stage biotech and digital health companies with innovative ideas and transformative new products and technologies of strategic interest to Sanofi. Among these areas are rare diseases, vaccines, potential cures in other core areas of Sanofi’s business footprint, and digital health solutions. For more information, visit www.sanofiventures.com.
About Magellan Health
Magellan Health, Inc., a Fortune 500 company, is a leader in managing the fastest growing, most complex areas of health, including special populations, complete pharmacy benefits and other specialty areas of healthcare. Magellan supports innovative ways of accessing better health through technology, while remaining focused on the critical personal relationships that are necessary to achieve a healthy, vibrant life. Magellan's customers include health plans and other managed care organizations, employers, labor unions, various military and governmental agencies and third-party administrators. For more information, visit MagellanHealth.com.
About Click Therapeutics
Click Therapeutics, Inc. develops and commercializes software as prescription medical treatments for people with unmet medical needs. Through cognitive and neurobehavioral mechanisms, Click’s Digital Therapeutics™ enable change within individuals, and are designed to be used independently or in conjunction with biomedical treatments. The Clickometrics® adaptive data science platform continuously personalizes user experience to optimize engagement and outcomes. Following a groundbreaking clinical trial, Click’s industry-leading smoking cessation program is available nationwide through a wide variety of payers, providers, and employers. Click’s lead prescription program is entering into a multi-center, randomized, controlled, parallel-group, phase III FDA registration trial for the treatment of Major Depressive Disorder in adults. For more information, visit ClickTherapeutics.com.
# # #
Company Contact
Sarah Jackson
Chief of Staff
sarah@clicktherapeutics.com
Media Contact
Stefanie Tuck
MacDougall Biomedical Communications
stuck@macbiocom.com
781-235-3060
Novel Enzyme Replacement Therapy in Development for Treatment of ENPP1 Deficiency
Boston, Mass., July 17, 2018 – Inozyme Pharma (Inozyme), a biopharmaceutical company dedicated to developing treatments for rare and debilitating diseases, today announced that both the Food and Drug Administration’s (FDA) Office of Orphan Products Development and the European Medicines Agency’s (EMA) Committee for Orphan Medical Products (COMP) granted Orphan Drug Designation to INZ-701 for the treatment of ENPP1 Deficiency. INZ-701, the Company’s lead therapeutic candidate, is in pre-clinical development for the treatment of patients with ENPP1 Deficiency, a serious and life-threatening calcification disorder that manifests as generalized arterial calcification of infancy (GACI) in infants and as autosomal recessive hypophosphatemic rickets type 2 (ARHR2) post-infancy.
“Orphan Drug Designation, both in the United States and the European Union, is an important regulatory milestone for Inozyme as we continue our quest to develop INZ-701 for patients with rare and life-threatening calcification disorders,” said Axel Bolte, co-founder and chief executive officer of Inozyme. “The dual designations from the FDA and EMA provide crucial momentum for INZ-701, putting us in an excellent position to rapidly advance the clinical development program for this novel enzyme replacement therapy.”
The FDA and EMA respectively grant Orphan Drug Designation to drugs intended for safe and effective treatment of rare, life-threatening or chronically debilitating conditions that affect fewer than 200,000 people in the United States or fewer than one in 2,000 individuals in Europe. By receiving Orphan Drug Designation, Inozyme qualifies for certain regulatory and financial incentives, including scientific assistance, fee reductions, tax credits and seven years of market exclusivity in the U.S., as well as 10 years of market exclusivity post-authorization in the European Union.
About ENPP1 Deficiency
The ENPP1 gene produces a critical enzyme called ectonucleotide pyrophosphatase/ phosphodiesterase 1 (ENPP1), which regulates inorganic pyrophosphate (PPi) levels in plasma. PPi is essential for preventing harmful soft tissue calcification and for regulating normal bone mineralization. ENPP1 Deficiency manifests as either generalized arterial calcification of infancy (GACI) type 1 or autosomal recessive hypophosphatemic rickets type 2 (ARHR2). GACI type 1 is a devastating and often fatal disease affecting infants and is characterized by calcification and narrowing of large and medium-sized arteries, resulting in heart failure and death in about half of patients within the first six months of life. ARHR2 manifests in the post-infancy stage and causes rickets, weakened bones, repeated bone fractures, skeletal deformities, short stature, muscle weakness, fatigue, and bone pain.
About INZ-701
INZ-701 is an enzyme replacement therapy under development with the intention to be used for the treatment of calcification disorders of the circulatory system, bones, and kidneys. In pre-clinical studies, the experimental therapy has shown potential to generate plasma pyrophosphate (PPi) and to restore it to appropriate physiological levels, thereby preventing calcification in the vasculature and kidneys and normalizing bone.
Inozyme is developing INZ-701 for certain rare, life-threatening and devastating genetic disorders such as ENPP1 Deficiency (GACI and ARHR2) and pseudoxanthoma elasticum (PXE) in which PPi levels are below the normal physiological levels. For more information about INZ-701, please visit: http://www.inozyme.com/our-science/.
About Inozyme Pharma
Inozyme Pharma is a biotechnology company committed to developing novel medicines for the treatment of rare metabolic diseases of calcification. The company was founded in 2016 with technology licensed from Yale University. For more information, please visit: www.inozyme.com.
Contact:
Inozyme Pharma
Henric Bjarke, COO
(617) 299-8321
henric.bjarke@inozyme.com
SmithSolve
Alex Van Rees
(973) 442-1555 ext. 111
alex.vanrees@smithsolve.com
NV‐5138 is a specific and direct activator of mTORC1, a cellular pathway required for the efficacy of many rapid acting antidepressants
CAMBRIDGE, Mass., June 26, 2018 – Navitor Pharmaceuticals, Inc., a biopharmaceutical company targeting the mTORC1 pathway to develop novel therapeutics that help patients live longer and healthier lives, announced today the initiation of a Phase 1 clinical study with its lead pipeline candidate, NV‐5138, for treatment‐resistant depression (TRD). NV‐5138 is a novel small molecule that directly activates mTORC1, a master cellular regulator that has recently been shown to be a central signaling pathway required for the efficacy of several rapid acting antidepressants. NV‐5138 is initially being evaluated in TRD but may offer future potential for the treatment in the broader disease category of major depressive disorder (MDD.)
“We are enthusiastic about initiating clinical development with NV‐5138 for major depressive disorder, as we believe this novel activator of mTORC1 has the potential to offer a unique approach to meeting many of the unmet needs of this serious and chronic disease. Millions of patients with depression do not adequately respond to standard pharmacological therapies which can take weeks or months before patients experience their effects, if at all, “said George P. Vlasuk, PhD, President and Chief Executive Officer of Navitor. “We see the development of NV‐5138 in MDD/TRD as a pioneering advance toward realizing the therapeutic potential of modulating the mTORC1 signaling pathway to treat a wide range of chronic human diseases.”
The Phase 1, multicenter, two‐part, double‐blind, placebo‐controlled study will evaluate the safety, tolerability and pharmacokinetics of NV‐5138 in up to 88 subjects, including healthy volunteers and patients diagnosed with TRD. In Part A, the single‐ascending‐dose portion of the study, up to 48 healthy volunteers will be randomly assigned to double‐blind treatment in six dosage‐level cohorts. Within each cohort, six subjects will be randomized to receive NV‐5138 and two subjects will be randomized to receive placebo. In Part B of the study, approximately 40 subjects diagnosed with TRD will be randomly assigned to double‐blind treatment at a single dosage level that will be established based on data from Part A of the study. Other prespecified outcome measures to be evaluated in Part B include standard depression rating and symptomology scores such as the Montgomery‐Åsberg Depression Rating Scale (MADRS).
“Initiation of this clinical study is supported by preclinical studies demonstrating the potential of NV‐5138 as an oral treatment for depression through activation of mTORC1, a cellular pathway that appears to underlie the beneficial effects of several in a new class of rapidly acting antidepressants,” said Maurizio Fava, MD, Director of the Division of Clinical Research of the Massachusetts General Hospital (MGH) Research Institute and member of the Navitor Clinical Advisory Board.
Previously, Navitor has presented preclinical results on the efficacy of NV‐5138 in multiple models of depression‐like behavior, which demonstrated that NV‐5138 produced behavioral responses and concomitant increases in new synapses (synaptogenesis) consistent with a rapid‐acting antidepressant through transient, direct activation of the mTORC1 signaling pathway in the brain. Navitor leveraged multiple preclinical observations that have shown mTORC1 activation is required for the efficacy of many rapid‐acting antidepressant compounds including several modulators of the NMDA (N‐methyl‐D‐aspartic acid)‐mediated signaling pathway like ketamine, which is an active area of innovative drug development for depression.
About Treatment Resistant Depression and Treatment Options
Treatment‐resistant depression (TRD) is a subset of major depressive disorder (MDD) that refers to depressive episodes that are not adequately controlled by standard antidepressant therapy. Several studies including a postmortem analysis of healthy and severely depressed patients as well as multiple pre‐clinical settings have suggested an association between the activity of mTORC1 pathway signaling and depression. Standard antidepressant therapies, such as selective serotonin reuptake inhibitors (SSRIs) and serotonin and
norepinephrine reuptake inhibitors (SNRIs) are only modestly effective and have a very slow onset typically taking 6‐8 weeks to show efficacy. Newer drugs that antagonize or otherwise modulate the presynaptic glutamate N‐methyl‐D‐aspartic acid (NMDA) receptor, have demonstrated the potential for improved efficacy with a rapid onset of antidepressant effects (days as opposed to weeks) and today there are several NMDA modulators in clinical development for depression, including ketamine and related agents. Unfortunately, presynaptic NMDA receptor modulation can cause significant side effects including dissociation (hallucination) and has abuse potential.
About Rapid Acting Antidepressants and mTORC1 Activity
New antidepressant drugs that modulate the presynaptic glutamate N‐methyl‐D‐aspartic acid (NMDA) receptor, have demonstrated the potential for improved efficacy with a rapid onset of antidepressant effects (days as opposed to weeks) and today there are several NMDA modulators in clinical development for depression, including ketamine and related agents. Since the initial observations connecting NMDA receptor modulation and depression, scientists have demonstrated that these agents increase production of key synaptic signaling proteins resulting in synaptogenesis and have also elucidated the mechanism that underlies the therapeutic antidepressant benefit seen with these agents in specific pre‐clinical settings. This new research demonstrates that presynaptic NMDA receptor modulation transiently activates the postsynaptic mTORC1 signaling pathway and this activation is required to initiate the cellular processes like protein synthesis that lead to the synaptogenesis and antidepressant effects of these agents. Although the initial target engagement of these agents occurs within a short time frame of a few hours, this transient activation of mTORC1 results in sustained, long‐lasting synaptic and behavioral effects that persist for days to even weeks after a single treatment.
About NV‐5138
NV‐5138 is an orally bioavailable, small molecule that is designed to directly and transiently activate mTORC1 activity by binding to and modulating a newly discovered cellular sensor protein for the amino acid leucine, which is a potent natural activator of mTORC1. Unlike leucine, oral administration of NV‐5138 results in significant mTORC1 pathway activation in the brain since it is not broken down or incorporated into new proteins. These properties make NV‐5138 a unique agent with which to evaluate the role of mTORC1 in brain disorders, such as depression, where mTORC1 activity is often suppressed. Results from preclinical models demonstrate that NV‐5138 produces rapid upregulation of key synaptic proteins, synaptogenesis and sustained antidepressant behavioral responses via the transient and direct activation of the mTORC1 signaling pathway. Since NV‐5138 does not directly modulate the NMDA receptor pathway, it may not have the side effects and abuse potential observed with several NMDA receptor therapeutics currently in development. NV‐ 5138 is currently being clinically studied for the treatment of major depressive disorder (MDD) with an initial focus on treatment‐resistant depression (TRD).
About Navitor
Navitor Pharmaceuticals, Inc. is realizing the potential of modulating mTORC1, the master regulator of cellular function, to develop a pipeline of therapeutics that help patients live longer and healthier lives. Our industry leading team is unlocking the promise of recent discoveries in mTORC1 biology to address a broad range of chronic diseases. Our initial clinical application is a first‐in‐class drug to address unmet needs in depression. For more information, please visit www.navitorpharma.com.
- Duman, RS and Aghajanian, GK. Science. 2012 October 5; 338(6103): 68–72.
- Scheung, L, et al., Frontiers in Neuroscience. 2015 July 21; 9 (249).
Contact:
The Yates Network
Kathryn Morris, 914‐204‐6412
kathryn@theyatesnetwork.com
CAMBRIDGE, Mass., June 25, 2018 (GLOBE NEWSWIRE) -- Unum Therapeutics Inc. (Nasdaq:UMRX), a clinical-stage biopharmaceutical company focused on the development of cellular immunotherapies based on its novel, universal Antibody-Coupled T cell Receptor (ACTR) technology platform, today announced the company’s addition to the Russell 3000 Index following the Russell US Indices’ annual reconstitution.
“We are pleased with our addition to the Russell 3000 Index, a leading US equity benchmark for institutional investors, which will further increase our exposure to these key investors,” said Christiana Stamoulis, President and Chief Financial Officer of Unum Therapeutics.
Membership in the Russell 3000 Index, which remains in place for one year, means automatic inclusion in the applicable growth and value style indexes. FTSE Russell determines membership for its Russell US Indexes primarily by objective, market capitalization rankings and style attributes. Russell US Indexes are widely used by investment managers and institutional investors as the basis for index funds and as benchmarks for active investment strategies. Approximately $9 trillion in assets are benchmarked against Russell US Indexes. Russell US Indexes are part of FTSE Russell, a leading global index provider.
About Unum Therapeutics
Unum Therapeutics is a clinical-stage biopharmaceutical company focused on the development and commercialization of novel immunotherapy products designed to harness the power of a patient’s immune system to cure cancer. Unum’s novel proprietary technology, antibody-coupled T cell receptor (ACTR), is a universal, engineered cell therapy intended to be used in combination with a wide range of tumor-specific antibodies to target different tumor types. Unum is actively building a pipeline of product candidates composed of ACTR T cells co-administered with antibodies for use in both hematologic and solid tumor cancers. The Company is headquartered in Cambridge, MA.
Forward-looking Statements
This press release contains forward-looking statements. Statements in this press release about our future expectations, plans and prospects, including projections regarding future revenues and financing performance, our long-term growth, the anticipated timing of our clinical trials and regulatory filings, the development of our product candidates, including the four lead ACTR product candidates, as well as other statements containing the words "anticipate," "believe," "continue," "could," "estimate," "expect," "intend," "may," "might," "plan," "potential," "predict," "project," "should," "target," "will," or "would" and similar expressions, constitute forward-looking statements within the meaning of the safe harbor provisions of The Private Securities Litigation Reform Act of 1995. We may not actually achieve the forecasts disclosed in our forward-looking statements, and you should not place undue reliance on our forward-looking statements. Actual results could differ materially from the projections disclosed in the forward-looking statements we make as a result of a variety of risks and uncertainties, including risks related to the accuracy of our estimates regarding expenses, future revenues, capital requirements, and the need for additional financing, the success, cost and timing of our product development activities and clinical trials, our ability to obtain and maintain regulatory approval for our product candidates, and the other risks and uncertainties described in the "Risk Factors" sections of our public filings with the Securities and Exchange Commission. In addition, the forward-looking statements included in this press release represent our views as of the date hereof. We anticipate that subsequent events and developments may cause our views to change. However, while we may elect to update these forward-looking statements at some point in the future, we specifically disclaim any obligation to do so. These forwardlooking statements should not be relied upon as representing our views as of any date subsequent to the date hereof.
Investor Contact:
Stern Investor Relations, Inc.
Mary T. Conway, 212-362-1200
mary@sternir.com
Media Contact:
Paul Kidwell, 617-680-1088
paul.kidwell@unumrx.com
- G100 directly targets and modifies TLR4 expressing malignant B cells making them more visible to the anti-tumor immune response-
SEATTLE and SOUTH SAN FRANCISCO, Calif., June 25, 2018 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), an immunotherapy company focused on novel therapies in oncology, today announced preclinical and translational data that support the mechanism of action of G100 in patients with indolent non-Hodgkin Follicular lymphomas (FL). These data were presented at the Inaugural AACR International Meeting Advances in Malignant Lymphoma: Maximizing the Basic-Translational Interface for Clinical Application 2018 in Boston.
The research presented was designed to understand why high TLR 4 expression in patient’s samples correlated with clinical responses to G100 treatment. By analyzing patient samples, cell lines and mouse lymphoma models the following was observed:
- Murine and human B-lymphoma cell lines express TLR4 and respond in vitro to G100 stimulation with upregulation of MHC-II and co-stimulatory markers CD40 and CD80, typical of the activation of antigen-presenting function of B-cells;
- In vivo murine tumors of lymphoma models respond to treatment with G100 in injected tumors as well as distal, untreated tumors showing local and abscopal tumor control, mediated by systemic T-cell response;
- Approximately 70% of follicular lymphoma patients in a Phase 1/2 study express TLR4 in >50% of tumor cells in baseline biopsies. TLR4 expression ranging from 10%-100% of tumor cells was also detected in biopsies of patients with marginal zone lymphoma, small lymphocytic lymphoma, diffuse large B-cell lymphoma and cutaneous T-cell lymphoma; and
- In an ongoing Phase 2 trial of G100 with low dose radiation and pembrolizumab, almost all patients with an objective tumor response (³50% tumor shrinkage) showed TLR4 expression in >50% of tumor cells.
“These data illustrate that in addition to the known activation by G100 of dendritic cells and macrophages in the tumor microenvironment, G100 can also act directly on malignant B cells expressing TLR4. G100 treated malignant B cells may become more visible to the anti-tumor immune response, which correlates with clinical responses following intratumoral therapy with G100.” said Jan ter Meulen, MD, PhD, Chief Scientific Officer at Immune Design. “In FL patients, a strong correlation was observed between expression of TLR4 in more than 50% of tumor cells and objective responses following G100 therapy. This discovery potentially allows for a TLR4 biomarker-targeted G100 therapy of other tumor types, independent of histology.”
The full poster presentation can be accessed from the publications page of the Immune Design website.
About G100
G100 is a product candidate from Immune Design's internal discovery platforms and contains a potent synthetic small molecule toll-like receptor-4 (TLR-4) agonist, Glucopyranosyl Lipid A (GLA). G100 leverages the activation of both innate and adaptive immunity in the tumor microenvironment to create an immune response against the tumor's preexisting diverse set of antigens. A growing set of clinical and preclinical data have demonstrated the ability of G100 to activate tumor-infiltrating lymphocytes, macrophages and dendritic cells, and promote antigen-presentation and the recruitment of T cells to the tumor. The ensuing induction of local and systemic immune responses has been shown to result in local and abscopal (shrinking of tumors outside the scope of the localized treatment) tumor control. G100 was evaluated in a Phase 1 study in Merkel cell carcinoma patients and produced a 50% overall response rate per protocol and a favorable safety profile. Currently, G100 is being evaluated as both a monotherapy (with local radiation) and in combination with Merck’s anti-PD-1 agent, pembrolizumab, pursuant to a clinical collaboration with Merck, in a randomized Phase 1/2 trial in patients with follicular non-Hodgkin lymphoma.
About Immune Design
Immune Design is a late-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic immune cells to fight cancer and other chronic diseases. CMB305 and G100, the leading product candidates with broad potential in oncology, are based on the company’s two technology platforms that are potent stimulators of the immune system – ZVex ® and GLAAS® - the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Both ZVex and GLAAS also have potential applications in infectious disease and allergy indications, which are being developed through ongoing pharmaceutical collaborations. Immune Design has offices in Seattle and South San Francisco. For more information, please visit www.immunedesign.com.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the timing of initiation, progress, scope and outcome of clinical trials for Immune Design's product candidates and the reporting of clinical data regarding Immune Design’s product candidates . Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, failure of Immune Design's collaborators to support or advance collaborations or product candidates and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Media Contact
Julie Rathbun
julie.rathbun@immunedesign.com
206-769-9219
Investor Contact
Sylvia Wheeler
Sylvia.wheeler@immunedesign.com
Digital Therapeutics Pioneer Launches Comprehensive,
Personalized Solution for Obesity-Related Chronic Disease
San Francisco, CA (June 20, 2018) -- Building on the company’s pioneering approach to digital behavior change, Omada Health today announced the availability of new programs that will help individuals control type 2 diabetes and hypertension. Adding to Omada’s industry-leading diabetes prevention program, the company will now offer integrated condition management programs for enterprise and health plan customers, significantly expanding the populations Omada serves. Leveraging seven years of data-driven insights; integration experience with hundreds of health plan and employer customers; and a proven approach to sustainable lifestyle change, Omada will now provide personalized interventions for those at-risk for, or with, certain obesity-related chronic conditions.
“Since the early days of the company, we’ve developed Omada to be an adaptable program that delivers meaningful health outcomes for participants, and return on investment for customers,” said Omada Health CEO Sean Duffy. “Today’s announcement is the next step in that journey. Clients, partners, and participants want a holistic digital healthcare provider built on clinical evidence, best-in-class user experience, and validated results. As we deliver new programs and features, Omada will continue to help our customers manage the health of their populations, as well as their healthcare spending.”
In addition to the new condition management programs, Omada is leveraging industry-leading data science and expert coaching to support participants in new ways - adding features for medication adherence and integrating remote monitoring for glucose and blood pressure. The company will continue to contract as a digital healthcare provider, filing medical claims with billing tied to participation and outcomes. The Omada program will adapt to each participant based on his or her:
- Demographic profile;
- Clinical profile, including comorbidities and multiple conditions;
- Self-identified personal barriers to success;
- Individual actions once enrolled in the program; and
- Use of connected glucometer and blood pressure cuff
"Omada delivers evidence-based behavior change by combining digitally-enabled coaching and clinical fidelity with sophisticated data science." said Omada VP of Medical Affairs Carolyn Bradner Jasik, MD, "We will now bring that expertise to the challenging areas of medication adherence and remote monitoring through an extensible, flexible platform that treats the whole person -- not just a single piece of their diagnosis."
A recent industry report by Willis Towers Watson illustrated the employer need, as 76 percent surveyed planned to invest in specific clinical solutions to diabetes to improve member health and reduce costs in 2019. The same survey reported 84 percent of companies will seek to identify and manage population chronic conditions across their workforce. Recent estimates place the total cost of diabetes at $327 billion annually , and high blood pressure at $131 billion per year.
About Omada Health
Omada is a digital intensive behavioral counseling program focused on reducing costly chronic disease in employer and health plan populations. The company offers a scalable, adaptable intervention for employees or members at risk for, or with, obesity-related chronic conditions such as type 2 diabetes and hypertension. Omada is the largest CDC-recognized provider of the National Diabetes Prevention Program, with deep integration experience with leading health plans, outcomes-based pricing, and ten peer-reviewed studies demonstrating the company’s ability to deliver lasting, clinically-meaningful results. To learn more, visit www.omadahealth.com .
Contact Information:
Adam Brickman
(914) 548-3748
Press@Omadahealth.com
CAMBRIDGE, Mass., June 13, 2018 – Inozyme Pharma (Inozyme), a biopharmaceutical company dedicated to developing treatments for rare and debilitating diseases, today introduced a no-cost, third-party genetic testing program designed to improve detection and understanding of two rare calcification disorders. Offered globally and in partnership with PreventionGenetics, the Inozyme program tests eligible participants for mutations in the ENPP1 and ABCC6 genes. Both of these genes are implicated in rare, severe calcification disorders, known as ENPP1 deficiency and ABCC6 deficiency, respectively, (sometimes called generalized arterial calcification of infancy [GACI] and autosomal recessive hypophosphatemic rickets type 2 [ARHR2]).
“The genetic testing program introduced today will help to enhance our understanding of ENPP1 deficiency and ABCC6 deficiency, with the ultimate goal of improving diagnosis and developing effective treatments,” said Axel Bolte, co-founder and chief executive officer of Inozyme. “We urge people with a family history of calcification disorders and their physicians to seek more information about the genetic testing program. Over time and through ongoing research, we hope to alleviate the life-limiting and life-threatening impact of these severe diseases.”
Inozyme created the genetic testing program to increase disease awareness, reduce barriers to genetic testing, and help people and their healthcare providers make more informed decisions about these rare conditions.
“The Inozyme genetic testing program will provide an early diagnostic testing measure for ENPP1 and ABCC6 deficiencies, which are indicators of serious calcification disorders that sometimes take years to diagnose accurately,” said James Weber, president of PreventionGenetics. “Access to reliable genetic testing may help shorten the diagnostic journey for patients, potentially lifting a significant emotional burden and paving the way for more timely and effective intervention. We are excited to work with Inozyme as we make the test available to physicians and their patients.”
The no-cost program offered by Inozyme and PreventionGenetics provides genetic testing to those who qualify. Although genetic testing can confirm a suspected diagnosis of a calcification disorder linked to ENPP1 or ABCC6 gene mutations, the absence of a genetic alteration does not preclude diagnosis of such a disease. For more information about the genetic testing program, please visit: www.inozyme.com/genetic-testing/.
About ENPP1 Deficiency
The ENPP1 gene produces a critical enzyme called ectonucleotide pyrophosphatase/ phosphodiesterase 1 (ENPP1), which regulates inorganic pyrophosphate (PPi) levels in plasma. PPi is essential for preventing harmful soft tissue calcification and for regulating normal bone mineralization. ENPP1 deficiency manifests as either generalized arterial calcification of infancy (GACI) type 1 or autosomal recessive hypophosphatemic rickets type 2 (ARHR2). GACI type 1 is a devastating and often fatal disease affecting infants and is characterized by calcification and narrowing of large and medium-sized arteries, resulting in heart failure and death in about half of patients within the first six months of life. ARHR2 usually manifests in the post-infancy stage, though it can occur in patients without prior GACI. ARHR2 causes rickets, weakened bones, repeated bone fractures, skeletal deformities, short stature, muscle weakness, fatigue and bone pain.
About ABCC6 Deficiency
Defects in the ABCC6 gene (ATP-binding cassette sub-family C member 6) lead to a decrease in plasma PPi and consequently to soft tissue calcification, and in rare circumstances cause GACI type 2 in infants. GACI type 2 is clinically similar to GACI type 1 and is also characterized by calcification and narrowing of large and medium-sized arteries, resulting in heart failure and death in about half of patients within the first six months of life.
About Inozyme Pharma
Inozyme Pharma is a biotechnology company committed to developing novel medicines for the treatment of rare diseases characterized by mineral imbalances, which lead to over-calcification of soft tissues and under-mineralization of bone. The company was founded in 2016 with technology licensed from Yale University. For more information, please visit: www.inozyme.com.
About PreventionGenetics
PreventionGenetics is a CLIA and ISO 15189:2012 accredited clinical DNA testing laboratory founded in 2004 and located in Marshfield, Wisconsin. PreventionGenetics provides patients with sequencing and deletion/duplication tests for nearly all clinically relevant genes, including whole exome sequencing, PGxomeⓇ.
Contact:
Inozyme Pharma
Henric Bjarke, COO
(617) 299-8321 henric.bjarke@inozyme.com
SmithSolve
Alex Van Rees
973-442-1555 ext. 111 alex.vanrees@smithsolve.com
-- 1st global Phase 3 trial focused on synovial sarcoma patients
SYNOVATE is a randomized, global Phase 3 trial to evaluate CMB305 immuontherapy in synovial sarcoma patients. www.synovatestudy.com
SEATTLE and SOUTH SAN FRANCISCO, Calif., May 24, 2018 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), an immunotherapy company focused on novel therapies in oncology, today announced the launch of the patient and healthcare provider websites for the SYNOVATE study – a pivotal trial to evaluate CMB305 immunotherapy in synovial sarcoma patients. Clinical sites will soon be open for enrollment initially in the US, with information available at www.synovatestudy.com.
“Patients with advanced synovial sarcoma often have few systemic treatment options immediately after they complete first line chemotherapy. Clinically, we often give them a break from therapy, following closely with the hope that they will not progress in the relatively short time period that is unfortunately common in this disease,” said William D. Tap, M.D., Medical Oncology and Chief, Sarcoma Medical Oncology Service, Memorial Sloan Kettering Cancer Center. “It is our hope that CMB305 may offer a new option for patients to receive a potentially beneficial therapy that will defer the need for subsequent therapy.”
SYNOVATE is a randomized, global Phase 3 clinical trial that will evaluate CMB305 monotherapy versus placebo in 248 patients 12 years of age and older with NY-ESO-1 positive, unresectable, locally-advanced or metastatic synovial sarcoma. Patients who are responding to a first-line therapy can become eligible for SYNOVATE following completion of their chemotherapy. SYNOVATE will be opened and recruiting patients in cancer centers throughout the United States, Canada, Europe, and the Asia Pacific region.
“We are very pleased to be working with high quality clinical groups around the country and internationally to launch the SYNOVATE study to explore CMB305 immunotherapy in patients with synovial sarcoma,” said Sergey Yurasov, M.D., Ph.D., Chief Medical Officer, Immune Design. “SYNOVATE is the first randomized, global Phase 3 trial of an immunotherapy focused in synovial sarcoma, and we hope it results in a new, approved therapy for these patients.”
Patient and Health Care Provider Resouces for SYNOVATE Study
Information can be found on the SYNOVATE website at www.synovatestudy.com and on the NIH clinical trial registry www.clinicaltrials.gov (Identifier: NCT03520959).
About Synovial Sarcoma
Soft tissue sarcomas are malignancies that arise from the soft tissues of the body, such as tissues that connect, support and surround other body structures including muscle, fat, blood vessels, nerves, tendons and the lining of joints. Synovial sarcoma is a sub type of soft tissue sarcoma where 70% of diagnoses occur in patients under 40 years old, is associated with a high risk of recurrence, and has been shown to have high expression of the NY-ESO-1 tumor antigen. The primary treatment for patients with locally advanced, unresectable or metastatic synovial sarcoma typically consists of an anthracycline-based chemotherapy regimen administered alone or in combination with other agents. Following disease progression after first line systemic therapy, treatment options are limited and median overall survival rates have been reported to be approximately 12 months. In connection with the planned Phase 3 study for CMB305 monotherapy, the FDA has agreed with Immune Design that synovial sarcoma patients constitute an unmet medical need.
About CMB305
CMB305 is a prime-boost cancer vaccine targeting NY-ESO-1-expressing tumors. NY-ESO-1 is a cellular protein that is typically found only in certain cancer cells and is often frequently expressed in synovial sarcomas. CMB305 is administered to participants by injection and is designed to provide clinical benefit by generating an anti-NY-ESO-1 immune response by activating a participant’s antigen-presenting dendritic cells.
About Immune Design
Immune Design is a late-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic immune cells to fight cancer and other chronic diseases. CMB305 and G100, the leading product candidates with broad potential in oncology, are based on the company’s two technology platforms that are potent stimulators of the immune system – ZVex® and GLAAS® - the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Both ZVex and GLAAS also have potential applications in infectious disease and allergy indications, which are being developed through ongoing pharmaceutical collaborations. Immune Design has offices in Seattle and South San Francisco. For more information, please visit www.immunedesign.com.
Cautionary Note on Forward-looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as “may,” “will,” “expect,” “plan,” “anticipate,” “target,” “estimate,” “intend” and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design’s expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties that could cause Immune Design’s clinical development programs, future results or performance to differ significantly from those expressed or implied by the forward-looking statements. Forwardlooking statements contained in this press release include, but are not limited to, statements about the progress, timing, scope and results of clinical trials and the timing and likelihood of obtaining regulatory approval of Immune Design’s product candidates. Many factors may cause differences between current expectations and actual results, including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, the uncertainties and timing of the regulatory approval process, and unexpected litigation or other disputes. Other factors that may cause Immune Design’s actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design’s filings with the U.S. Securities and Exchange Commission, including the “Risk Factors” sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Media Contact
Julie Rathbun
julie.rathbun@immunedesign.com
206-769-9219
Investor Contact
Sylvia Wheeler
sylvia.wheeler@immunedesign.com
650-392-8318.
April 3, 2018
CAMBRIDGE, Mass., April 03, 2018 (GLOBE NEWSWIRE) -- Unum Therapeutics Inc. (“Unum Therapeutics”) (NASDAQ:UMRX), a clinical-stage biopharmaceutical company focused on the development of novel immunotherapy products designed to harness the power of a patient’s immune system to cure cancer, today announced the closing of its initial public offering of 5,770,000 shares of common stock at a public offering price of $12.00 per share. The gross proceeds from this offering, before deducting underwriting discounts and commissions and other offering expenses, are approximately $69.2 million. Unum Therapeutics’ common stock began trading on the Nasdaq Global Select Market under the ticker symbol "UMRX" on March 29, 2018.
Morgan Stanley and Cowen are acting as joint book-running managers for the initial public offering. SunTrust Robinson Humphrey and Wedbush PacGrow are acting as lead managers.
In addition to the shares sold in the initial public offering, Unum Therapeutics announced the concurrent sale of an additional 416,666 shares at the public offering price of $12.00, for gross proceeds of $5.0 million, in a private placement to Seattle Genetics, Inc., an existing shareholder of Unum Therapeutics. The sale of these shares of common stock is not registered under the Securities Act of 1933, as amended, and the shares are subject to a 180-day lock-up agreement.
A registration statement relating to the securities issued in the initial public offering was declared effective by the Securities and Exchange Commission (the “SEC”) on March 28, 2018. The offering will be made only by means of a prospectus. Copies of the final prospectus related to the offering may be obtained by visiting EDGAR on the SEC Web site at www.sec.gov or from Morgan Stanley & Co. LLC, Attention: Prospectus Department, 180 Varick Street, 2nd Floor, New York, New York 10014, or Cowen and Company, LLC, c/o Broadridge Financial Services, 1155 Long Island Avenue, Edgewood, NY 11717, Attention: Prospectus Department, or by calling (631) 274-2806.
This press release shall not constitute an offer to sell or the solicitation of an offer to buy these securities, nor shall there be any sale of these securities in any state or jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction.
About Unum Therapeutics
Unum Therapeutics uses its proprietary antibody-coupled T cell receptor (ACTR) technology in combination with tumor-targeting antibodies to activate the body’s own immune system to fight cancer. Unum Therapeutics is actively building a pipeline of ACTR programs in combination with a wide range of proprietary, tumortargeting antibodies for use in both hematologic and solid tumor cancers. The Company is headquartered in Cambridge, MA.
Contact:
Unum Therapeutics Inc.
Christiana Stamoulis, +1-617-843-5352
christiana.stamoulis@unumrx.com
Source: Unum Therapeutics Inc.
– Names Kevin M. Forrest, Ph.D. President and CEO
– Proprietary technology based on pioneering work from the lab of Matthew D. Disney, Ph.D. of The Scripps Research Institute Florida
San Diego, January 3, 2018 -- Expansion Therapeutics, Inc., a new 5AM Ventures formed private company focused on the discovery and development of ribonucleic acid (RNA) targeted small molecule medicines, announced today the close of a $55.3 million Series A financing co-led by 5AM Ventures, Kleiner Perkins, Novartis Venture Fund, and Sanofi Ventures with participation from RA Capital Management and Alexandria Venture Investments. Proceeds will advance Expansion’s portfolio of small molecule drugs targeting key human disease-driving RNAs with an initial focus on expansion repeat disorders, a set of approximately 30 genetic diseases that currently have no satisfactory treatments.
“In a short period of time Expansion has assembled a leading team, the key scientific founder in Matt Disney of The Scripps Research Institute Florida, and a capital efficient plan to advance the emerging field of RNA targeted small molecule medicines,” said Scott M. Rocklage, Ph.D., Managing Partner of 5AM Ventures and founding investor and Chairman of the Board of Directors of Expansion Therapeutics. “We look forward to continuing to work with the company to develop medicines for patients with few treatment options.”
“The science underpinning Expansion’s RNA targeted medicines offers great promise in the treatment of a set of RNA triggered diseases, such as myotonic dystrophy, that currently have no viable therapies. We are excited to invest in a company that could make a real difference to patients suffering from these incurable diseases,” said Beth Seidenberg, M.D., General Partner of Kleiner Perkins.
Coincident with the close of the Series A, Expansion co-founder Kevin M. Forrest, Ph.D., was named president and chief executive officer. Dr. Forrest previously served as founding chief operating officer and chief financial officer for the San Diego-based anti-infectives company Cidara Therapeutics (Nasdaq: CDTX). Prior, he was a Principal at 5AM Ventures. Dr. Forrest holds a B.S. in biology from Boston College and a Ph.D. in molecular biology from Princeton University where he published on various RNA regulatory processes.
RNA Targeted Small Molecule Medicines
Incubated within 5AM Ventures’ 4:59 Initiative, and subsequently seed funded by 5AM and Sanofi Ventures, Expansion’s approach is based on key patent-protected platform technologies and enabling tools pioneered in the laboratory of Dr. Disney of The Scripps Research Institute, who is the leader in the field of small molecule targeting of RNA.
“I am gratified that our efforts over the past dozen years have culminated in this important opportunity,” said Dr. Disney. “It is clear that disease-related RNA is now an addressable target with small molecule medicines and we are now on the verge of developing treatments for patients with the most urgent medical needs. We will work tirelessly to fulfill this promise.”
Expansion repeat disorders include myotonic dystrophy type I (DM1), which is the leading cause of adult onset muscular dystrophy. “Expansion repeat disorders, in particular DM1, represent an attractive first application of our technology as it is well established that toxic RNA drives disease,” said Dr. Forrest. “Furthermore, our small molecule approach has the potential to address both peripheral and central symptoms that are debilitating for patients.”
Expansion Names Board of Directors and Forms Scientific Advisory Board
Following the close of the financing, the Expansion board of directors will include:
- Scott M. Rocklage, Ph.D., of 5AM Ventures and Chairman of the Expansion Board of Directors
- Matthew D. Disney, Ph.D., of The Scripps Research Institute
- Kevin M. Forrest, Ph.D., of Expansion Therapeutics
- Jason P. Hafler, Ph.D., of Sanofi Ventures
- Yujiro S. Hata, M.B.A., of Ideaya Biosciences
- Campbell Murray, M.D., M.B.A., M.P.P., of Novartis Venture Fund
- Beth Seidenberg, M.D., of Kleiner Perkins
- Andrew Levin, M.D., Ph.D., of RA Capital Management joins as a board observer
Expansion has also formed a scientific advisory board comprised of leaders in the field of RNA targeted small molecule chemistry and biology, RNA folding, and structural biology, including:
- Robert T. Batey, Ph.D., of the University Colorado, Boulder
- Dale L. Boger, Ph.D., of The Scripps Research Institute
- Ronald R. Breaker, Ph.D., of the Howard Hughes Medical Institute and Yale University
- M.G. Finn, Ph.D., of the Georgia Institute of Technology
- David H. Mathews, M.D., Ph.D., of the University of Rochester
- Michael Zuker, Ph.D., of the Rensselaer Polytechnic Institute
About RNA Targeted Small Molecule Medicines
Ribonucleic acid, or RNA, is a biomolecule that was once thought to be a simple messenger between DNA, or deoxyribonucleic acid, and protein. Recent advances in biology, however, have shown that RNA plays a much greater role than previously appreciated. This includes control of gene expression via long non-coding RNAs, RNA stability via small interfering RNAs, RNA translation via transfer RNAs and microRNAs, and even cellular communication (RNA exosomes). RNA can form higher order structures that form the basis of “druggable” surfaces and pockets that can be targeted by small molecule therapeutics. The best examples of RNA targeted small molecule medicines include several FDA approved classes of antibiotics that bind and inactivate key structures in bacterial RNAs.
About Myotonic Dystrophy Type I
Myotonic dystrophy type I (DM1) affects at least 1 in 8,000 people, or 40,000 individuals in the U.S. alone, and is the most frequent cause of adult onset muscular dystrophy. DM1 is caused by a toxic expansion in RNA, which leads to multi-systemic symptoms including muscular, cardiac, respiratory, gastrointestinal, endocrine, and central nervous system defects. This genetic disease often affects entire families, with progressively worsening disease across generations, and there are no effective treatment options available for DM1.
For more information about myotonic dystrophy type I, visit www.myotonic.org.
About Expansion Therapeutics, Inc.
Expansion Therapeutics is a drug discovery and development company pursuing the vast potential of small molecule medicines for RNA-mediated diseases. Based on exclusive worldwide rights to groundbreaking research from the laboratory of Matthew D. Disney, Ph.D., at The Scripps Research Institute, Expansion has assembled the intellectual property, know-how, and proprietary enabling technologies and tools necessary to facilitate the creation of potent and specific small molecule binders of RNA. Through this unique platform, Expansion is building a portfolio of novel RNA-targeted drug candidates with activity across a broad number of disease indications. The company’s initial development focus is on therapies for patients with expansion repeat diseases who currently have limited and unsatisfactory treatment options. Expansion is based in San Diego, California and Jupiter, Florida.
For more information, visit www.expansionrx.com.
Media Contact:
Christy Curran
Sam Brown Inc.
christycurran@sambrown.com
615.414.8668
NEW YORK, Dec. 19, 2017 (GLOBE NEWSWIRE) -- Ovid Therapeutics Inc. (NASDAQ:OVID), a biopharmaceutical company committed to developing medicines that transform the lives of people with rare neurological diseases, today announced that the U.S. Food and Drug Administration (FDA) has granted Fast Track designation to OV101 for the treatment of Angelman syndrome.
“This designation is an important milestone for both Ovid and the Angelman community as it enables increased dialog with the FDA, speeding our ability to bring this potential therapeutic option to people living with Angelman syndrome. We believe that OV101, with its novel mechanism of action, has the potential to be an innovative and impactful therapy,” said Amit Rakhit, M.D., MBA, chief medical and portfolio management officer of Ovid Therapeutics. “In addition to the regulatory milestones of orphan drug and Fast Track designations for Angelman syndrome, we achieved significant clinical progress with our OV101 program. As a result of positive Phase 1 data, we were recently able to expand our ongoing Phase 2 STARS clinical trial to include both adults and adolescents with Angelman syndrome. We look forward to data from the STARS trial in the second half of 2018.”
OV101 is a delta (δ)-selective GABAA receptor agonist that targets the disruption of tonic inhibition, a central physiological process of the brain that is thought to be the underlying cause of Angelman syndrome and other neurodevelopmental disorders. Ovid is currently studying OV101 in its Phase 2 STARS clinical trial, a randomized, double-blind, placebo-controlled study investigating the safety and efficacy of OV101 in patients with Angelman syndrome. Upon successful completion of a Phase 1 pharmacokinetic (PK) and safety study showing that OV101 has a similar PK profile in adolescents as in adults, Ovid recently amended the STARS protocol to include patients aged 13 years and older.
The FDA’s Fast Track process is designed to expedite the development and review of drugs used to treat serious conditions and fill an unmet medical need. Fast Track designation enables the company to have early and frequent communication with the FDA throughout the drug development and review process, often leading to faster drug approval and patient access.
About OV101
OV101 (gaboxadol) is believed to be the only delta (δ)-selective GABAA receptor agonist in development and the first investigational drug to specifically target the disruption of tonic inhibition, a central physiological process of the brain that is thought to be the underlying cause of certain neurodevelopmental disorders. OV101 has been demonstrated in laboratory studies and animal models to selectively activate the δ-subunit of GABAA receptors, which are found in the extrasynaptic space (outside of the synapse), and thereby impact neuronal activity through tonic inhibition.
Ovid is developing OV101 for the treatment of Angelman syndrome and Fragile X syndrome to potentially restore tonic inhibition and relieve several of the symptoms of these disorders. In preclinical studies, it was observed that OV101 improved symptoms of Angelman syndrome and Fragile X syndrome. Gaboxadol has previously been tested in over 4,000 patients (approximately 950 patient-years of exposure) and was observed to have favorable safety and bioavailability profiles.
The FDA has granted orphan drug and Fast Track designations for OV101 for the treatment of Angelman syndrome and orphan drug designation for the treatment of Fragile X syndrome. The U.S. Patent and Trademark Office has granted Ovid patents directed to methods of treating Angelman syndrome using OV101. The issued patents expire in 2035 for Angelman syndrome.
About Angelman Syndrome
Angelman syndrome is a genetic disorder that is characterized by a variety of signs and symptoms. Characteristic features of this disorder include delayed development, intellectual disability, severe speech impairment, problems with movement and balance, seizures, sleep disorders and anxiety. The most common cause of Angelman syndrome is the disruption of a gene that codes for ubiquitin protein ligase E3A (UBE3A). Angelman syndrome affects approximately 1 in 12,000 to 20,000 people in the U.S. There are currently no U.S. Food and Drug Administration (FDA)-approved therapies for the treatment of Angelman syndrome.
Angelman syndrome is associated with a reduction in tonic inhibition, a function of the delta (δ)-selective GABAA receptor that allows a human brain to decipher excitatory and inhibitory neurological signals correctly without being overloaded. If tonic inhibition is reduced, the brain becomes inundated with signals and loses the ability to separate background noise from critical information.
About Ovid Therapeutics
Ovid Therapeutics (NASDAQ:OVID) is a New York-based biopharmaceutical company using its BoldMedicine™ approach to develop therapies that transform the lives of patients with rare neurological disorders. Ovid’s drug candidate, OV101, is currently in development for the treatment of Angelman syndrome and Fragile X syndrome. Ovid initiated the Phase 2 STARS trial of OV101 in people with Angelman syndrome in 2017 and completed a Phase 1 trial in adolescents with Angelman syndrome or Fragile X syndrome. Ovid is also developing OV935 in collaboration with Takeda Pharmaceutical Company Limited for the treatment of epileptic encephalopathies and in August 2017 initiated a Phase 1b/2a trial of OV935.
For more information on Ovid, please visit http://www.ovidrx.com/.
Forward-Looking Statements
This press release includes certain disclosures that contain “forward-looking statements,” including, without limitation, statements regarding progress, timing, scope and results of clinical trials for Ovid’s product candidates, the timing of clinical data, the development of therapies for younger patients, the provision of access to effective therapies, and the FDA Fast Track process leading to faster drug approval and patient access. You can identify forward-looking statements because they contain words such as “will,” “believes” and “expects.” Forward-looking statements are based on Ovid’s current expectations and assumptions. Because forward-looking statements relate to the future, they are subject to inherent uncertainties, risks and changes in circumstances that may differ materially from those contemplated by the forward-looking statements, which are neither statements of historical fact nor guarantees or assurances of future performance. Important factors that could cause actual results to differ materially from those in the forward-looking statements are set forth in Ovid’s filings with the Securities and Exchange Commission, including its Quarterly Report on Form 10-Q for the quarter ended June 30, 2017, under the caption “Risk Factors.” Ovid assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Contacts
Investors:
Burns McClellan
Steve Klass, 212-213-0006
Sklass@burnsmc.com
Media:
Pure Communications, Inc.
Katie Engleman, 910-509-3977
katie@purecommunicationsinc.com
Industry Experts Join Curisium’s Inaugural Advisory Board
Manhattan Beach, CA - December 14, 2017 - Curisium, a healthcare technology and services company that enables innovative contracting at scale via its blockchain-based platform, announced today it has raised $3.5M in seed financing from Flare Capital Partners, New Enterprise Associates (NEA), Shuttle Fund, Sanofi Ventures, and Green Bay Ventures. In conjunction with the financing, Bill Geary of Flare Capital and Mohamad Makhzoumi of NEA have joined the Curisium Board of Directors, and Ruchita Sinha of Sanofi Ventures has been appointed a Board Observer.
The Curisium platform uses cutting-edge blockchain and secure computation technologies to allow payers, providers, and life science companies to efficiently and securely engage in innovative, patient-centric value-based contracts. Nearly a third of the payments in the $3T US healthcare market are already tied to some form of innovative payment model; Curisium aims to transform healthcare by disrupting existing frameworks and enabling scalable value-based care contracting at the individual patient level.
“Payers, providers, and life science companies are increasingly entering into various forms of innovative contracts,” said Peter Kim, co-founder and CEO of Curisium. “However, effective implementation today is hampered by costly logistics, lack of trust, and difficulty verifying patient-level outcomes.”
“Curisium’s platform, by enabling outcome verification at the patient level, while automating the payment side, has the potential to rapidly accelerate the breadth and depth of innovative contracting arrangements,” said Bill Geary, co-founder and Partner at Flare Capital. “We’re thrilled to be an investor partner with the Curisium founding team having successfully backed them before, and are impressed with their deep industry and technology insights and capabilities.”
“There is no mechanism today to effectively define or share patient health states,” added Milind Kamkolkar, Chief Data Officer at Sanofi. “With the Curisium platform, we now have a trusted version of health records that will get better over time, and the industry can finally start to discover, maintain and improve patient health.”
Greg Papadopoulos, Venture Partner at NEA and former CTO of Sun Microsystems, noted “Healthcare data are siloed, and there is a reluctance to share due to fundamental distrust among its custodians. Curisium’s platform tackles this logjam with unique cryptographic guarantees around its data access.”
Both Kamkolkar and Papadopoulos have joined Curisium’s inaugural Industry Advisory Board.
“Blockchain technology has tremendous potential and there is a huge opportunity to implement this application within the healthcare industry,” stated Mohamad Makhzoumi, General Partner at NEA and Head of Healthcare Services. “We are thrilled to partner with the Curisium team as they pioneer blockchain for healthcare in a pragmatic, market-driven way, that addresses pain points across trust, security, and payment logistics.”
To learn more about Curisium, please visit www.curisium.com.
About Curisium
Curisium is a healthcare technology and services company based in Manhattan Beach, CA that provides scalable innovative contracting solutions with its blockchain-based platform. Founded by seasoned healthcare technology executives, Curisium provides tailored solutions for payers, providers, and life science companies to enter into patient-centric, secure and efficient innovative contracting arrangements. For more information, please visit www.curisium.com.
About Flare Capital Partners
Flare Capital is a team of proven healthcare technology venture capital investors known for their strategic industry resources, insight and total commitment to the success of its entrepreneurs. Flare Capital raised one of the healthcare industry’s largest dedicated venture capital funds focused exclusively on early stage and emerging growth investments in healthcare technology innovation and is privileged to closely partner with founders and management. Investments include Aetion, Bright Health, Circulation, ClearDATA, Curisium, Evolent Health, HealthReveal, HealthVerity, Iora Health, VisitPay and Welltok. Learn more at www.flarecapital.com.
About NEA
New Enterprise Associates, Inc. (NEA) is a global venture capital firm focused on helping entrepreneurs build transformational businesses across multiple stages, sectors and geographies. With more than $20 billion in cumulative committed capital since the firm’s founding in 1977, NEA invests in technology and healthcare companies at all stages in a company’s lifecycle, from seed stage through IPO. The firm's long track record of successful investing includes more than 210 portfolio company IPOs and more than 360 acquisitions. For additional information, visit www.nea.com.
About Shuttle Fund
Shuttle is a global investment fund focused on disruptive blockchain-based technologies. Decentralized blockchain technologies are redefining the way people and business interact, and Shuttle has a successful track record of investing into and advising fundamentally disruptive blockchain-based platforms across multiple domains. For more information, visit www.shuttlefund.com.
About Sanofi Ventures
Sanofi Ventures is the corporate venture capital arm of Sanofi. Sanofi Ventures invests in early-stage biotech and digital health companies with innovative ideas and transformative new products and technologies that are of strategic interest to Sanofi. Among these areas are rare diseases, vaccines, potential cures in other core areas of Sanofi’s business footprint, and digital health solutions. For more information, visit www.sanofiventures.com.
About Green Bay Ventures
Green Bay Ventures (GBV) is a San Francisco based venture capital firm investing at the intersection of technology and large markets including manufacturing, energy, transportation, logistics, real estate and telecommunications. GBV focuses on a small number of high conviction opportunities and works side-by-side with a select group of visionary entrepreneurs to create breakthrough technologies and transform industries. For more information, visit www.greenbayventures.com.
NEW YORK, Dec. 05, 2017 (GLOBE NEWSWIRE) -- Ovid Therapeutics Inc. (NASDAQ:OVID), a biopharmaceutical company committed to developing medicines that transform the lives of people with rare neurological diseases, today announced that the U.S. Food and Drug Administration (FDA) has granted orphan drug designation to TAK-935/OV935 for the treatment of Dravet syndrome, a severe and rare form of childhood epilepsy that typically presents during the first year of life. Takeda Pharmaceutical Company Limited and Ovid formed a global collaboration to develop and commercialize TAK-935/OV935 for the treatment of developmental and epileptic encephalopathies in January 2017.
Dravet syndrome is classified as a developmental and epileptic encephalopathy, a group of rare epilepsies that cause significant morbidities and can worsen over time. Children with Dravet syndrome experience frequent seizures, loss of muscle control, cognitive deficits and, in approximately 10 percent of cases, death before the age of 12 years. Moreover, in those who survive into adulthood, their long-term intellectual development and seizure outcomes are typically extremely poor.
“We are pleased by the FDA’s decision to grant orphan drug designation to TAK-935/OV935 for the treatment of Dravet syndrome, a severe and debilitating disease,” said Dr. Emiliangelo Ratti, head of Takeda’s Neuroscience Therapeutic Area Unit. “This designation is a significant step forward in researching a potential treatment option for people living with Dravet syndrome for whom therapeutic options are severely limited, and an important milestone for this investigational molecule.”
TAK-935/OV935 is a potent, highly-selective, first-in-class inhibitor of the enzyme cholesterol 24-hydroxylase (CH24H). It is believed that CH24H is involved in over-activation of the glutamatergic pathway, which has been shown to play a role in the initiation and spread of seizure activity. To Ovid and Takeda’s knowledge, TAK-935/OV935 is the only molecule with this mechanism of action in clinical development.
“We believe that TAK-935/OV935, with its novel mechanism of action, has the potential to be an innovative treatment for people with rare epilepsies, such as Dravet syndrome,” said Matthew During M.D., DSc, FACP, FRACP, president and chief scientific officer of Ovid Therapeutics. “We have rapidly advanced this program into a Phase 1b/2a clinical trial and anticipate data in 2018. We look forward to continuing our work with Takeda to bring this potentially transformative therapy to patients.”
Orphan drug designation is intended to facilitate and expedite drug development for rare diseases for which there are no current treatments available. It also provides substantial benefits to the sponsor, including the potential for tax credits for clinical development costs, study-design assistance, and several years of market exclusivity for the product upon regulatory approval.
About Dravet Syndrome
Dravet syndrome is a severe form of childhood epilepsy that typically presents during the first year of life. It is believed to be largely caused by mutations in the SCN1A gene. Children experience frequent seizures, loss of muscle control, cognitive deficits and, in approximately 10 percent of cases, death before the age of 12 years. While some patients may survive into adulthood, their long-term intellectual development and seizure outcomes are typically extremely poor. The incidence of Dravet syndrome in the United States ranges from 1 in 15,700 to 1 in 20,900 births. Patients are frequently treated with combinations of classic anti-epileptic drugs, none of which are particularly effective. No drugs have been approved specifically for the treatment of Dravet syndrome in the United States and only one drug, the anticonvulsant stiripentol, has been approved in Europe.
Dravet syndrome is one of several disorders which together are designated as developmental and epileptic encephalopathies. This group includes epilepsy syndromes associated with severe cognitive and behavioral disturbances. The International League Against Epilepsy (ILAE) defines an epileptic encephalopathy as a condition in which “the epileptiform EEG abnormalities themselves are believed to contribute to a progressive disturbance in cerebral function.”
These epilepsies cause significant morbidities for patients beyond what might be expected from the known underlying pathology alone and can worsen over time. Developmental and epileptic encephalopathies typically present early in life and are often associated with severe cognitive and developmental impairment in addition to frequent treatment-resistant seizures throughout the person’s lifetime. These disorders vary in age of onset, developmental outcomes, etiologies, neuropsychological deficits, electroencephalographic (EEG) patterns, seizure types and prognosis.
About TAK-935/OV935
TAK-935/OV935, which is being studied in developmental and epileptic encephalopathies, is a potent, highly-selective, first-in-class inhibitor of the enzyme cholesterol 24-hydroxylase (CH24H). CH24H is predominantly expressed in the brain, where it plays a central role in cholesterol homeostasis. CH24H converts cholesterol to 24S-hydroxycholesterol (24HC), which then exits the brain into the blood plasma circulation. Glutamate is one of the main neurotransmitters in the brain and has been shown to play a role in the initiation and spread of seizure activity. Recent literature indicates 24HC is involved in over-activation of the glutamatergic pathway through modulation of the NMDA channel, implying its potential role in central nervous system diseases such as epilepsy. To Ovid and Takeda’s knowledge, TAK-935/OV935 is the only molecule with this mechanism of action in clinical development.
TAK-935/OV935 has been tested in preclinical models to provide data to support the advancement of the drug into human clinical studies in patients suffering from rare epilepsy syndromes. A novel proprietary PET ligand, developed by Takeda and Molecular Neuroimaging, LLC (MNI), has been used to determine target enzyme occupancy of TAK-935/OV935 in the brain. In addition, the effect of TAK-935/OV935 on CH24H enzyme activity in the brain has been assessed by following measurable reductions in the plasma concentration of 24HC.
TAK-935/OV935 has completed four Phase 1 clinical studies, which have assessed tolerability and target engagement at doses believed to be therapeutically relevant. TAK-935/OV935 is being co-developed by Ovid and Takeda Pharmaceutical Company Limited.
About Ovid Therapeutics
Ovid Therapeutics (NASDAQ:OVID) is a New York-based biopharmaceutical company using its BoldMedicine™ approach to develop therapies that transform the lives of patients with rare neurological disorders. Ovid’s drug candidate, OV101, is currently in development for the treatment of Angelman syndrome and Fragile X syndrome. Ovid initiated the Phase 2 STARS trial of OV101 in people with Angelman syndrome in 2017 and completed a Phase 1 trial in adolescents with Angelman syndrome or Fragile X syndrome. Ovid is also developing OV935 in collaboration with Takeda Pharmaceutical Company Limited for the treatment of epileptic encephalopathies and in August 2017 initiated a Phase 1b/2a trial of OV935.
For more information on Ovid, please visit http://www.ovidrx.com/.
Forward-Looking Statements
This press release includes certain disclosures that contain “forward-looking statements,” including, without limitation, statements regarding the progress, timing, scope and results of clinical trials for Ovid’s product candidates, the reporting of clinical data regarding Ovid’s product candidates, and the potential use of TAK-935/OV935 to treat rare epilepsies. You can identify forward-looking statements because they contain words such as “will,” “believes” and “expects.” Forward-looking statements are based on Ovid’s current expectations and assumptions. Because forward-looking statements relate to the future, they are subject to inherent uncertainties, risks and changes in circumstances that may differ materially from those contemplated by the forward-looking statements, which are neither statements of historical fact nor guarantees or assurances of future performance. Important factors that could cause actual results to differ materially from those in the forward-looking statements are set forth in Ovid’s filings with the Securities and Exchange Commission, including its Quarterly Report on Form 10-Q for the quarter ended September 30, 2017, under the caption “Risk Factors.” Ovid assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Contacts
Investors:
Burns McClellan
Steve Klass, 212-213-0006
Sklass@burnsmc.com
Media:
Pure Communications, Inc.
Katie Engleman, 910-509-3977
katie@purecommunicationsinc.com
Company bolsters leadership team with several executive appointments
Cambridge, Mass., November 15, 2017 - Inozyme Pharma, Inc., a biotechnology company developing novel medicines to treat rare diseases of calcification, affecting soft tissues and bone, today announced it has raised a $49 million Series A financing. The financing was led by Longitude Capital, and included participation from New Enterprise Associates (NEA), Novo Ventures and Sanofi Ventures.
“Our mission is to develop potentially disease-modifying therapies to help children who are affected with rare, but severe and debilitating disorders of metabolism. These patients have very poor treatment options,” said Axel Bolte, chief executive officer and co-founder of Inozyme Pharma. “We have attracted a premier syndicate of healthcare investors who are committed to helping us achieve our goal, and this funding positions us well to advance our therapeutic approach.”
Inozyme Pharma was founded in 2016 with technology developed in the laboratory of Demetrios Braddock, M.D., Ph.D., and licensed from Yale University. The company will use the proceeds from this financing to advance its lead enzyme replacement therapy for the treatment of Generalized Arterial Calcification of Infancy (GACI) and Autosomal Recessive Hypophosphatemic Rickets Type 2 (ARHR2) into the clinic. These disorders are characterized by mineral imbalances that lead to over calcification of soft tissues and under mineralization of bone.
“Inozyme Pharma’s deep understanding of the biology of calcification will be used to develop new medicines that have the potential to drastically improve the standard-of-care,” said Reinaldo Diaz, venture partner at Longitude Capital, and a member of Inozyme Pharma’s board of directors. “With strong foundational intellectual property and a multidisciplinary team of experts, the company is well-positioned to advance new therapies for underserved populations.”
In addition to the Series A financing, Inozyme Pharma has also expanded its experienced leadership team of industry veterans, including:
- Axel Bolte, president, chief executive officer, and co-founder Axel Bolte brings 20 years of experience in biotech and healthcare venture capital, with considerable background in rare diseases, to Inozyme Pharma. Mr. Bolte has served as a board member of various private and public companies.
- Henric Bjarke, senior vice president and chief operating officer Henric Bjarke’s 20 years in pharma and biotech bring a wealth of experience in metabolic and rare diseases to Inozyme Pharma. Having previously served as vice president and therapeutic area head for the metabolic business unit at Alexion Pharmaceuticals, Mr. Bjarke was responsible for asfotase-alfa for the treatment hypophosphatasia and other development programs.
- Steven Jungles, senior vice president and chief technical operations officer Steven Jungles offers 20 years of industry experience to the Inozyme Pharma leadership team. Before joining Inozyme Pharma, Mr. Jungles served as senior vice president, technical operations at Ultragenyx Pharmaceutical, vice president of contract manufacturing and supply chain at BioMarin Pharmaceuticals and worked at the Harvard Gene Therapy Initiative.
- Eric Yuen, senior vice president and chief medical officer Erin Yuen brings 24 years of experience in academia and the biopharmaceutical industry. Dr. Yuen has developed biologics and small molecules for a variety of diseases, including rare genetic, neurology, psychiatry, pain, and oncology disorders. Dr. Yuen has held several senior positions at Merck, Johnson & Johnson, Ultragenyx and BioClin.
- Ruhi Ahmed, vice president of regulatory and government affairs ; Ruhi Ahmed’s 15 years of experience in regulatory affairs include drug development and life cycle management of programs from the preclinical to the commercial stage. Prior to Inozyme Pharma, Dr. Ahmed held positions at Ultragenyx and BioMarin Pharmaceuticals where she worked on the MPS, XLH and PKU programs.
- Stephen Basso, vice president of finance Stephen Basso has 25 years of experience in life sciences and financial services that he brings to Inozyme Pharma. Prior to joining the company, Mr. Basso was vice president of corporate finance at Alexion, where he supported the development and commercialization of Soliris, Strensiq and Kanuma.
In addition, Inozyme has appointed the following seasoned directors to its board:
- Joseph Schlessinger, chairman and co-founder, Yale University • Reinaldo Diaz, Longitude Capital
- Ed Mathers, New Enterprise Associates
- Martin Edwards, Novo Holdings A/S
- Axel Bolte, president, chief executive officer and co-founder
About GACI and ARHR2
GACI is an ultra-rare, autosomal recessive orphan disease affecting infants. It is caused by loss-of-function mutations in either the ENPP1 or ABCC6 gene, and results in low circulating levels of pyrophosphate (PPi) and calcification of medium and large arteries and heart. GACI presents as a crisis within the first week of life and is associated with high mortality rates. The majority of patients are likely to die within the first year of life. If affected children survive past six months, they are likely to continue living but develop ARHR2, a rare skeletal disorder characterized by low levels of serum PPi, which can result in rickets, repeated fractures of the long bones, rachitic skeletal deformities, and impaired growth and development.
About Inozyme Pharma, Inc.
Inozyme Pharma is a biotechnology company committed to developing novel medicines for the treatment of rare diseases characterized by mineral imbalances, which lead to over calcification of soft tissues and under mineralization of bone. The company was founded in 2016 with technology licensed from Yale University.
MEDIA CONTACT:
Lauren Barbiero
W2Opure
lbarbiero@w2ogroup.com
+1 (646) 564-2156
SEATTLE and SOUTH SAN FRANCISCO, Calif., Oct. 19, 2017 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on oncology, today announced that the European Medicines Agency (EMA) has granted Orphan Drug Designation for G100, Immune Design’s investigational intratumoral therapy, for the treatment of follicular non-Hodgkin’s lymphoma.
The EMA orphan drug designation is assigned to products targeting the treatment of rare diseases, which are defined as having a prevalence of not more than 5 in 10,000 people in the European Union (EU). This designation provides the sponsor with certain benefits, including protocol assistance, reduced fees for regulatory activities and up to 10 years of market exclusivity in the EU upon marketing approval for the designated indication.
G100 has also been granted orphan drug designation by the U.S. Food and Drug Administration for the treatment of follicular non-Hodgkin's lymphoma.
G100 is a product candidate from Immune Design's GLAAS® discovery platform. It contains a potent synthetic small molecule toll-like receptor-4 (TLR-4) agonist, Glucopyranosyl Lipid A (GLA), and is the lead product candidate in Immune Design’s Antigen Agnostic approach. G100 activates innate and adaptive immunity in the tumor microenvironment to generate an immune response against the tumor's preexisting diverse set of antigens. A growing set of clinical and preclinical data have demonstrated the ability of G100 to activate tumor-infiltrating lymphocytes, macrophages and dendritic cells, and promote antigen-presentation and the recruitment of T cells to the tumor. The induction of local and systemic immune responses has been shown in preclinical studies to result in local and abscopal (shrinking of tumors outside the scope of the localized treatment) tumor control. Currently, G100 is being evaluated as both a monotherapy (with local radiation) and in combination with Merck’s anti-PD-1 agent, pembrolizumab, pursuant to a clinical collaboration with Merck, in a randomized Phase 1/2 clinical trial in patients with follicular non-Hodgkin’s lymphoma.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the two leading product candidates focused in cancer immunotherapy, are the first products from Immune Design’s two separate discovery platforms targeting dendritic cells in vivo, ZVex® and GLAAS®. Both ZVex and GLAAS also have potential
applications in infectious disease and allergy as demonstrated by ongoing pharmaceutical collaborations. Immune Design has offices in Seattle and South San Francisco. For more information, please visit www.immunedesign.com.
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
NEW YORK, Oct. 16, 2017 (GLOBE NEWSWIRE) -- Ovid Therapeutics Inc. (NASDAQ:OVID), a biopharmaceutical company committed to developing medicines for patients with rare neurological diseases, today announced new positive preclinical data on OV101 that shows normalization of behavioral abnormalities that resemble those seen in people with Fragile X syndrome. The improvements seen with OV101, a novel agonist of the extrasynaptic GABA receptor, were consistent across multiple behavioral measures in a model of Fragile X syndrome. The data were presented at the 18th International Fragile X and Related Neurodevelopmental Disorders Workshop.
“Treatment with OV101 results in significant behavioral improvements that are consistent across all behavioral endpoints tested,” said Matthew During, M.D., DSc, FACP, FRACP, president and chief scientific officer of Ovid Therapeutics. “We designed this study to deepen our understanding of the mechanisms of action and potential benefit of OV101 to treat patients with Fragile X syndrome.”
In the study, researchers demonstrated that acute administration of 0.5 mg/kg of OV101 to Fmr1 knockout mice fully normalized behavioral abnormalities relevant to Fragile X syndrome (hyperactivity, anxiety, irritability and aggression, and restricted and repetitive behaviors). All effects were highly statistically significant (p < 0.001). It is believed that symptoms of Fragile X syndrome are a result of disrupted tonic inhibition, the key to the brain's ability to discriminate signal from noise. The results presented indicate that by specifically targeting the delta (δ)-subset of GABA receptors, OV101 may be able to alleviate symptoms of Fragile X syndrome by modulating the GABA pathway and restoring tonic inhibition.
“We have built a strong foundation in scientific analysis, translational medicine, drug development and regulatory capabilities. This study builds upon this foundation and underscores the potential role of OV101 in modulating tonic inhibition, an important underlying mechanism in certain neurodevelopmental disorders,” said Amit Rakhit M.D., MBA, chief medical and portfolio officer of Ovid Therapeutics. “Together with the recent FDA orphan drug designation for OV101 for the treatment of Fragile X syndrome, this data is another important step in our disciplined strategy to develop OV101 as a potential first in class compound for Fragile X syndrome.”
About OV101
OV101 (gaboxadol) is believed to be the only delta (δ)-selective GABAA receptor agonist in development and the first investigational drug to specifically target the disruption of tonic inhibition that is thought to be the underlying cause of certain neurodevelopmental disorders. OV101 has been demonstrated in laboratory studies and animal models to selectively activate the δ-subunit of GABAA receptors, which are found in the extrasynaptic space (outside of the synapse), and thereby impact neuronal activity through tonic inhibition.
Ovid is developing OV101 for the treatment of Angelman syndrome and Fragile X syndrome to potentially restore tonic inhibition and relieve several of the symptoms of these disorders. In preclinical studies, it was observed that OV101 improved symptoms of Angelman syndrome and Fragile X syndrome. To date, gaboxadol has been tested in over 4,000 patients (approximately 950 patient-years of exposure) and was observed to have favorable safety and bioavailability profiles.
The FDA granted orphan drug designation for OV101 for the treatment of both Angelman syndrome and Fragile X syndrome. The United States Patent and Trademark Office has granted Ovid two patents directed to methods of treating Angelman syndrome using OV101. The issued patents expire in 2035, without regulatory extensions.
About Fragile X Syndrome
Fragile X syndrome is the most common inherited form of intellectual disability and autism, with a prevalence of 1 in 3,600 to 4,000 males and 1 in 4,000 to 6,000 females in the United States. Individuals with Fragile X syndrome often have a range of behavioral challenges, such as cognitive impairment, anxiety, mood swings, hyperactivity, attention deficit, poor sleep, self-injury and heightened sensitivity to various stimuli, such as sound. Additionally, individuals with Fragile X syndrome are prone to comorbid medical issues including seizures and sleep disturbance. Fragile X syndrome results from mutations in the FMR1 gene, which blocks expression of the Fragile X Mental Retardation Protein (FMRP), an important protein in GABA synthesis. There are no FDA-approved therapies for Fragile X syndrome, and treatment primarily consists of behavioral interventions and pharmacologic management of symptoms.
In studies of individuals with Fragile X syndrome and in experimental models, extrasynaptic GABA levels are abnormally reduced, and there is also dysregulation of GABA receptors. This ultimately contributes to a decrease in tonic inhibition, causing the brain to become inundated with signals and lose the ability to separate background noise from critical information.
About Ovid Therapeutics
Ovid Therapeutics (NASDAQ:OVID) is a New York-based biopharmaceutical company using its BoldMedicine™ approach to develop therapies that transform the lives of patients with rare neurological disorders. Ovid’s drug candidate, OV101, is currently in development for the treatment of Angelman syndrome and Fragile X syndrome. Ovid has initiated the Phase 2 STARS trial of OV101 in adults with Angelman syndrome and a Phase 1 trial in adolescents with Angelman syndrome or Fragile X syndrome. Ovid is also developing OV935 in collaboration with Takeda Pharmaceutical Company Limited for the treatment of rare epileptic encephalopathies and has initiated a Phase 1b/2a trial of OV935.
For more information on Ovid, please visit http://www.ovidrx.com/.
Forward-Looking Statements
This press release includes certain disclosures that contain “forward-looking statements,” including, without limitation, statements regarding progress, timing, scope and results of clinical trials for Ovid’s product candidates, the reporting of clinical data regarding Ovid’s product candidates, the development of OV101 as a potential first in class compound for Fragile X syndrome and the potential use of TAK-935/OV935 to treat rare epilepsies. You can identify forward-looking statements because they contain words such as “will,” “believes” and “expects.” Forward-looking statements are based on Ovid’s current expectations and assumptions. Because forward-looking statements relate to the future, they are subject to inherent uncertainties, risks and changes in circumstances that may differ materially from those contemplated by the forward-looking statements, which are neither statements of historical fact nor guarantees or assurances of future performance. Important factors that could cause actual results to differ materially from those in the forward-looking statements are set forth in Ovid’s filings with the Securities and Exchange Commission, including its Quarterly Report on Form 10-Q for the quarter ended June 30, 2017, under the caption “Risk Factors.” Ovid assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Contacts
Investors:
Burns McClellan
Steve Klass, 212-213-0006
Sklass@burnsmc.com
Media:
Pure Communications, Inc.
Katie Engleman, 910-509-3977
katie@purecommunicationsinc.com
- CMB305 Monotherapy vs. Placebo in the Maintenance Setting
- PFS Analysis for Potential Full Approval as Early as 24 Months After Study Start
- Conference Call and Webcast Tomorrow at 5:30am PT/8:30am ET
SEATTLE and SOUTH SAN FRANCISCO, Calif., Oct. 16, 2017 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on oncology, today announced that based on productive discussions with the U.S. Food and Drug Administration (FDA), it plans to initiate a pivotal Phase 3 trial to support a Biologics License Application (BLA) for CMB305, a novel cancer vaccine, in patients with synovial sarcoma.
The randomized Phase 3 trial will evaluate CMB305 monotherapy vs. placebo in patients with NY-ESO-1+ locally advanced unresectable or metastatic synovial sarcoma, a sub type of soft tissue sarcoma, who have no evidence of progression after first-line chemotherapy. Immune Design intends to start the study in mid-2018 and enroll 248 patients aged twelve and older. Patients will be randomized 1:1 to receive either CMB305 monotherapy or placebo. The trial will have progression free survival (PFS) followed by overall survival (OS) as co-primary endpoints. If the PFS endpoint is successful, the FDA offered that it may support full approval of CMB305. Depending on the rate of events, final PFS analysis may occur as early as 24 months from the first patient dosed.
About Synovial Sarcoma
Soft tissue sarcomas are malignancies that arise from the soft tissues of the body, such as tissues that connect, support and surround other body structures including muscle, fat, blood vessels, nerves, tendons and the lining of joints. Synovial sarcoma is a sub type of soft tissue sarcoma where 70% of diagnoses occur in patients under 40 years old, is associated with a high risk of recurrence, and has been shown to have high expression of the NY-ESO-1 tumor antigen. The primary treatment for patients with locally advanced, unresectable or metastatic synovial sarcoma typically consists of an anthracycline-based chemotherapy regimen administered alone or in combination with other agents. Following disease progression after first line systemic therapy, treatment options are limited and median overall survival rates have been reported to be approximately 12 months. In connection with the planned Phase 3 study for CMB305 monotherapy, the FDA has agreed with Immune Design that synovial sarcoma patients constitute an unmet medical need.
About CMB305
CMB305 is an investigational prime-boost vaccine approach against NY-ESO-1-expressing tumors, designed to generate an integrated, anti-NY-ESO-1 immune response in vivo via a targeted, specific interaction with dendritic cells, a mechanism of action Immune Design believes differs from traditional cancer vaccines. CMB305 is being evaluated in soft tissue sarcoma patients in ongoing Phase 1 monotherapy and Phase 2 combination studies. Immune Design has received Orphan Drug Designation for CMB305 from the FDA for the treatment of soft tissue sarcoma, as well as from the FDA and European Commission for each of the components of CMB305 for the treatment of soft tissue sarcoma.
Conference Call Information
Immune Design will host a conference call and live audio webcast tomorrow, October 17, at 5:30 a.m. Pacific time / 8:30 a.m. Eastern time to discuss the CMB305 development strategy and pivotal trial design.
To participate in the conference call, please dial 844-266-9538 for domestic callers and 216-562-0391 for international callers and provide the conference ID 9299539, or access the listen-only live webcast by visiting the investor relations section of the company website at http://ir.immunedesign.com/events.cfm.
A telephone replay of the call will be available for five days by dialing 855-859-2056 for domestic callers or 404-537-3406 for international callers and entering the conference code: 9299539. An archived copy of the webcast will be available on Immune Design's website beginning approximately two hours after the conference call and will be available for at least 30 days after the conference call.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the two leading product candidates focused in cancer immunotherapy, are the first products from Immune Design’s two separate discovery platforms targeting dendritic cells in vivo, ZVex® and GLAAS®. Both ZVex and GLAAS also have potential applications in infectious disease and allergy as demonstrated by ongoing pharmaceutical collaborations. Immune Design has offices in Seattle and South San Francisco. For more information, please visit www.immunedesign.com.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the “safe harbor” provisions of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties that could cause our clinical development programs, future results or performance to differ significantly from those expressed or implied by the forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the design, progress, timing, scope and potential results of the pivotal Phase 3 clinical trial of CMB305 in synovial sarcoma patients, the possibility that PFS data will be sufficient to support regulatory approval and the timing of the PFS analysis. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data, clinical trial site activation or enrollment rates that are slower than expected, changes in expected or existing competition, changes in the regulatory environment, the uncertainties and timing of the regulatory approval process and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
- Phase 2 Interim Analysis Data Presented at ESMO 2017 Congress
SEATTLE and SOUTH SAN FRANCISCO, Calif., Sept. 08, 2017 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on oncology, today announced that an interim analysis of its ongoing, randomized Phase 2 trial showed that NY-ESO-1+ soft tissue sarcoma (STS) patients receiving the combination of CMB305 and Genentech’s checkpoint inhibitor, atezolizumab (TECENTRIQ®), experienced greater clinical benefit and immune response than those receiving atezolizumab alone. The data will be presented in a poster discussion session at the European Society of Medical Oncology (ESMO) 2017 Congress, September 8-12, 2017 in Madrid, Spain.
This fully enrolled trial is evaluating the safety, immunogenicity and efficacy of CMB305 in combination with atezolizumab (C+A, n=45), or atezolizumab alone (A, n=43), in a total of 88 patients with locally advanced, relapsed, or metastatic NY-ESO-1+ synovial sarcoma or myxoid/round-cell liposarcoma. Data to be presented at ESMO summarize patients evaluated in an interim analysis in a data cut as of July 21, 2017, with analysis divided into two groups: pre-specified interim analysis (n=36) and full study population (n=88).
Patient Characteristics:
Patients receiving CMB305 plus atezolizumab have more advanced disease than those receiving atezolizumab alone, including:
- Metastatic Disease: 98% (C+A) vs. 79% (A)
- ≥2 prior lines of systemic anti-cancer therapy: 61% (C+A) vs. 49% (A)
- ≥2 lesions at time of study entry: 96% (C+A) vs. 84% (A)
- Grade 3 disease at diagnosis: 47% (C+A) vs. 33% (A)
Greater Clinical Benefit with Combination of CMB305 and Atezolizumab:
Interim analysis data (n=36) show that patients receiving CMB305 plus atezolizumab experienced greater clinical benefit than those receiving atezolizumab alone.
- Disease Control Rate (Partial Responses (PR) + Stable Disease (SD)): 61%, including 1 PR (C+A) vs. 28% with no PRs (A)
- Median Progression Free Survival (PFS): 2.6 months (C+A) vs. 1.4 months (A)
- Time to Next Treatment (TTNT): 9 months (C+A) vs. 6.3 months (A)
- Overall survival: as of the collection date, overall survival data are immature (median duration of observation is less than six months); Immune Design intends to present survival data in 2018 once all patients approach at least one year of follow up.
In the full study population (n=88), the trend of greater clinical benefit on the combination arm remains consistent for the entire patient population:
- Disease Control Rate: 57% (3 PRs total, 1 unconfirmed) (C+A) vs. 38% (0 responses) (A)
More Robust Immune Response with Combination of CMB305 and Atezolizumab:
Patients in the full study population (n=88) who received the combination of CMB305 and atezolizumab demonstrated stronger anti-NY-ESO-1 immune responses compared to those receiving atezolizumab alone (samples evaluable from (n=60/88)), including:
- Induced anti-NY-ESO-1 T cells: 52% (C+A) vs. 17% (A)
- Induced anti-NY-ESO-1 antibodies: 52% (C+A) vs. 0% (A)
- Induced antigen spreading: 19% (C+A) vs. 0% (A)
Biomarker Analysis Shows Continued Link Between Induced Immune Response and Clinical Benefit:
In an exploratory analysis, a trend towards improved overall survival was observed in patients with an induced immune response (T cells or antibodies) who received CMB305 plus atezolizumab.
- Induced anti-NY-ESO-1 T cells: 78% reduction in mortality rate, as compared to patients without induced T cells [HR=0.22, log-rank p value=0.043]
- Induced anti-NY-ESO-1 antibodies: 87% reduction in mortality rate, as compared to patients without induced antibodies [HR=0.13, log-rank p value=0.025]
- This trend of improved overall survival in patients with induced immune response was not observed in the atezolizumab-only arm.
In addition, pretreatment tumor biopsies available from 70 patients show both an absent or very low level of PD-L1 expression and CD8 T cell infiltration, further supporting that these subtypes of STS are "cold" tumors.
Positive Safety Profile with Combination of CMB305 and Atezolizumab:
This combination was observed to be well tolerated, and there were no new safety signals in either arm.
“We believe the greater clinical benefit and more robust immune response shown in the combination study arm supports the hypothesis that the combination of an appropriately-designed, next-generation cancer vaccine such as CMB305 with a checkpoint inhibitor is important to produce clinical benefit in a cold tumor such as STS, where checkpoint inhibitors otherwise may have limited efficacy,” said Carlos Paya, M.D., Ph.D., President and Chief Executive Officer of Immune Design. “In addition, we are pleased to observe the trend towards longer overall survival in patients with an induced anti-NY-ESO-1 immune response, further supporting the positive CMB305 monotherapy data we presented at ASCO in June.”
Atezolizumab is a monoclonal antibody being developed by Genentech, a member of the Roche Group, and is designed to target and bind to a protein called PD-L1 (programmed death ligand-1). The trial is being conducted pursuant to a clinical collaboration with Genentech. TECENTRIQ® is a registered trademark of Genentech.
The ESMO Poster Discussion session presentation details are as follows:
A Phase 2 Study of CMB305 and Atezolizumab in NY-ESO-1+ Soft Tissue Sarcoma: Interim Analysis of Immunogenicity, Tumor Control and Survival
Abstract # 1480PD
Session Title: Sarcoma Poster Discussion Session
Date: Monday, Sept. 11, 2017
Time: 11 a.m. – 12:30 p.m.
Location: Bilbao Auditorium
Poster Presenter: Dr. Sant Chawla
Poster Discussant: Dr. Robert Maki
About CMB305
CMB305 is a prime-boost vaccine approach against NY-ESO-1-expressing tumors, designed to generate an integrated, anti-NY-ESO-1 immune response in vivo via a targeted, specific interaction with dendritic cells, a mechanism of action Immune Design believes differs from traditional cancer vaccines. CMB305 is being evaluated in soft tissue sarcoma patients in ongoing Phase 1 monotherapy and Phase 2 combination studies. Immune Design has received Orphan Drug Designation for CMB305 from the U.S. Food and Drug Administration (FDA) for the treatment of soft tissue sarcoma, as well as from the FDA and European Medicines Agency for each of the components of CMB305 for the treatment of soft tissue sarcoma.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the two leading product candidates focused in cancer immunotherapy, are the first products from Immune Design’s two separate discovery platforms targeting dendritic cells in vivo, ZVex® and GLAAS®. Both ZVex and GLAAS also have potential applications in infectious disease and allergy as demonstrated by ongoing pharmaceutical collaborations. Immune Design has offices in Seattle and South San Francisco. For more information, please visit www.immunedesign.com.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties that could cause our clinical development programs, future results or performance to differ significantly from those expressed or implied by the forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the progress, timing, scope and results of clinical trials, the association of data with treatment outcomes, and the timing and likelihood of obtaining regulatory approval of Immune Design's product candidates. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, the uncertainties and timing of the regulatory approval process, and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
NEW YORK, Sept. 06, 2017 (GLOBE NEWSWIRE) -- Ovid Therapeutics Inc. (NASDAQ:OVID), a biopharmaceutical company committed to developing medicines for patients with rare neurological disorders, today announced the appointment of Ana C. Ward as senior vice president and general counsel.
“With over 20 years of legal and bioscience operational experience, along with a deep understanding of how to achieve success within fast-growing life science companies, Ana will be a strong addition to our leadership team,” said Jeremy Levin, D.Phil., MB Chir, chairman of the board of directors and chief executive officer of Ovid. “Her perspective, breadth of experience, proven capability and expertise complements that of our team. We are pleased to welcome Ana to the Ovid team.”
“With two rapidly progressing clinical programs, this is an exciting time to join such a dynamic company,” said Ms. Ward. “In just a few short years, Ovid has already established a sound strategy to address an important area of need. I am excited to contribute to the continued growth and success of the company and work closely with its talented management team.”
Ms. Ward previously served as general counsel, executive vice president of patient access and corporate development, and board secretary of Rosetta Genomics, Ltd., overseeing public company reporting and executing licensing and partnership activities. Prior to Rosetta Genomics, Ms. Ward held a series of leadership roles within the Ambion family of companies, including Ambion, Inc. (acquired by Applied Biosystems), Asuragen, Inc. (a spin-off of Ambion, Inc.) and Mirna Therapeutics, Inc. (a spin-off of Asuragen, Inc.), where she was responsible for managing a diverse portfolio of legal matters such as intellectual property claims, compliance, trademark clearance, and strategic business interactions. Ms. Ward holds a bachelor of arts in French, a master of science in
molecular biology and a master of business administration, all from the University of Texas, Austin, and a juris doctorate from the University of Texas Law School. Ms. Ward also holds a master of science in bioscience regulatory affairs from Johns Hopkins University. She is admitted to practice law in the State of Texas and is registered to practice before the United States Patent & Trademark Office.
About Ovid Therapeutics
Ovid Therapeutics (NASDAQ: OVID) is a New York-based biopharmaceutical company using its BoldMedicine™ approach to develop therapies that transform the lives of patients with rare neurological disorders. Ovid’s drug candidate, OV101, is currently in development for the treatment of Angelman syndrome and Fragile X syndrome. Ovid has initiated the Phase 2 STARS trial of OV101 in adults with Angelman syndrome and a Phase 1 trial in adolescents with Angelman syndrome or Fragile X syndrome. Ovid is also developing OV935 in collaboration with Takeda Pharmaceutical Company Limited for the treatment of rare epileptic encephalopathies and has initiated a Phase 1b/2a trial of OV935.
For more information on Ovid, please visit http://www.ovidrx.com/.
Contacts
Investors:
Burns McClellan
Steve Klass, 212-213-0006
Sklass@burnsmc.com
Media:
Pure Communications, Inc.
Katie Engleman, 910-509-3977
katie@purecommunicationsinc.com
- Interim data analysis shows greater clinical benefit and immune response with CMB305+atezolizumab than with atezolizumab alone
SEATTLE and SOUTH SAN FRANCISCO, Calif., Aug. 30, 2017 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on oncology, today announced positive topline data from its interim analysis of the ongoing, randomized Phase 2 trial evaluating CMB305 in combination with atezolizumab (TECENTRIQ®) or atezolizumab alone in 88 soft tissue sarcoma patients. The data will be presented in a poster discussion session at the European Society of Medical Oncology (ESMO) 2017 Congress, September 8-12, 2017 in Madrid, Spain.
“The two main goals of this study are (1) to determine whether combining a next-generation vaccine like CMB305 with a checkpoint inhibitor (such as an anti-PD-L1 antibody) provides improved clinical benefit compared to that of the checkpoint inhibitor alone, particularly in a PD-L1-low or -negative tumor, and (2) in a randomized setting, to determine the biological activity of CMB305,” said Carlos Paya, M.D., Ph.D., President and Chief Executive Officer of Immune Design. “We believe these interim data constitute the first steps towards answering both questions in a favorable manner.”
Data to be presented at ESMO build upon those data on the first 36 patients summarized in the abstract, and include a greater number of patients enrolled.
- Clinical Benefit: Data from the larger patient population show that patients receiving CMB305 and atezolizumab experienced greater clinical benefit in the form of Disease Control Rate (including objective responses), Progression Free Survival and Time to Next Treatment than those receiving atezolizumab alone.
- Immune Response: Patients who received the combination of CMB305 and atezolizumab demonstrated an increased frequency of induced immune responses to NY-ESO-1, including NY-ESO-1-specific T cells, NY-ESO-1 antibodies, and antigen spreading, in comparison to patients who received atezolizumab alone
- Biomarkers: In an exploratory analysis, a trend towards improved overall survival was observed in patients in the CMB305 and atezolizumab combination arm who had pre-existing and treatment-induced anti-NY-ESO-1 immunity, compared to the atezolizumab alone arm. Pre-treatment tumor biopsy analysis showed negligible levels of PD-L1 expression.
- Safety: No new safety signals have been observed in either arm.
The fully enrolled trial is evaluating the safety, immunogenicity and efficacy of CMB305 in combination with atezolizumab, or atezolizumab alone, in a total of 88 patients with locally advanced, relapsed, or metastatic NY-ESO-1+ synovial sarcoma or myxoid/round-cell liposarcoma. Atezolizumab is a monoclonal antibody designed to target and bind to a protein called PD-L1 (programmed death ligand-1) and is being developed by Genentech, a member of the Roche Group. The trial is being conducted pursuant to a clinical collaboration with Genentech. Immune Design intends to present survival data in 2018 once all patients have at least one year of follow up (current data are preliminary: median duration of observation <six months).
The ESMO Poster Discussion session presentation details are as follows:
A Phase 2 Study of CMB305 and Atezolizumab in NY-ESO-1+ Soft Tissue Sarcoma: Interim Analysis of Immunogenicity, Tumor Control and Survival
Abstract # 1480PD
Session Title: Sarcoma Poster Discussion Session
Date: Monday, Sept. 11, 2017
Time: 11 a.m. – 12:30 p.m.
Location: Bilbao Auditorium
Poster Presenter: Dr. Sant Chawla
Poster Discussant: Dr. Robert Maki
About CMB305
CMB305 is a prime-boost vaccine approach against NY-ESO-1-expressing tumors, designed to generate an integrated, anti-NY-ESO-1 immune response in vivo via a targeted, specific interaction with dendritic cells, a mechanism of action Immune Design believes differs from traditional cancer vaccines. CMB305 is being evaluated in soft tissue sarcoma patients in ongoing Phase 1 monotherapy and 2 combination studies. Immune Design has received Orphan Drug Designation for CMB305 from the U.S. Food and Drug Administration (FDA) for the treatment of soft tissue sarcoma, as well as from the FDA and European Medicines Agency for each of the components of CMB305 for the treatment of soft tissue sarcoma.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigenspecific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the primary foci of Immune Design's ongoing immunooncology clinical programs, are products of its two synergistic discovery platforms, ZVex and GLAAS, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, please visit www.immunedesign.com.
TECENTRIQ® (atezolizumab) is a registered trademark of Genentech, a member of the Roche Group.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forwardlooking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties that could cause our clinical development programs, future results or performance to differ significantly from those expressed or implied by the forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the progress, timing, scope and results of clinical trials, the association of data with treatment outcomes, and the timing and likelihood of obtaining regulatory approval of Immune Design's product candidates. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, the uncertainties and timing of the regulatory approval process, and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
NEW YORK, June 23, 2017 (GLOBE NEWSWIRE) Ovid Therapeutics Inc. (NASDAQ:OVID), a biopharmaceutical company committed to developing medicines for patients with rare neurological disorders, today announced that it will be added to the Russell 2000®, 3000® and Microcap® Indexes, effective as of the close of the market on Friday, June 23, 2017, following Russell’s annual reconstitution of its comprehensive set of U.S. and global equity indexes.
Russell indexes are widely used by investment managers and institutional investors for index funds and as benchmarks for active investment strategies. Russell determines membership for its equity indexes primarily by objective, market capitalization rankings and style attributes. The Russell 2000 Index measures the performance of the small cap segment of the U.S. equity universe. The Russell 2000 Index is a subset of the Russell 3000 Index representing approximately 10 percent of the total market capitalization of that index. The Russell Microcap Index represents 2,000 small cap and microcap stocks and captures the smallest 1,000 companies in the Russell 2000, in addition to 1,000 smaller U.S.based listed stocks.
About Ovid Therapeutics
Ovid Therapeutics (NASDAQ:OVID) is a New York based, biopharmaceutical company using its BoldMedicineTM approach to develop therapies that transform the lives of patients with rare neurological disorders. Ovid’s drug candidate, OV101, is currently in development for the treatment of Angelman syndrome and Fragile X syndrome. Ovid has initiated the Phase 2 STARS trial of OV101 in adults with Angelman syndrome and a Phase 1 trial in adolescents with Angelman syndrome or Fragile X syndrome. Ovid is also developing OV935 in collaboration with Takeda Pharmaceutical Company Limited for the treatment of rare epileptic encephalopathies. Ovid expects to initiate a Phase 1b/2a trial of OV935 to treat rare epileptic encephalopathies in 2017.
For more information on Ovid, please visit http://www.ovidrx.com/.
Forward Looking Statements
This press release includes certain disclosures which contain “forward looking statements,” including, without limitation, statements regarding the scope and timing of initiating a Phase 1b/2a trial of OV935 to treat rare epileptic encephalopathies. You can identify forward looking statements because they contain words such as “will,” “believes” and “expects.” Forward looking statements are based on Ovid’s current expectations and assumptions. Because forward looking statements relate to the future, they are subject to inherent uncertainties, risks and changes in circumstances that may differ materially from those contemplated by the forward looking statements, which are neither statements of historical fact nor guarantees or assurances of future performance. Important factors that could cause actual results to differ materially from those in the forward looking statements are set forth in Ovid’s filings with the Securities and Exchange Commission, including its Quarterly Report on Form 10Q for the quarter ended March 31, 2017, under the caption “Risk Factors.” Ovid assumes no obligation to update any forward looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Ovid Contacts:
Investors:
Burns McClellan
Steve Klass, 212‐213‐0006
Sklass@burnsmc.com
Media:
Pure Communications, Inc.
Katie Engleman, 910‐509‐3977
katie@purecommunicationsinc.com
Data from CMB305 and G100 to be Presented at ASCO Annual Meeting in June 2017
SEATTLE and SOUTH SAN FRANCISCO, May 17, 2017 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinicalstage immunotherapy company focused on oncology, reported new clinical and biomarker data today from CMB305 and G100 monotherapy studies. The American Society of Clinical Oncology (ASCO) is publishing three abstracts today relating to these new data. A broader set of data will be presented at the ASCO 2017 Annual Meeting, providing further clinical validation of the company's lead product candidates and discovery platforms.
CMB305 Monotherapy in Patients with Soft Tissue Sarcoma
- l Most recent patient survival data meaningfully exceed published survival outcomes for standard of care therapy in comparable soft tissue sarcoma (STS) patients with recurrent metastatic disease.
1. With a median follow up exceeding 18 and 11 months for LV305 and CMB305, respectively, median overall survival (mOS) has not yet been reached in recurrent metastatic STS patients.
- Durable disease control was observed in more than half of STS patients, including durable tumor growth arrest in patients who had evidence of disease progression prior to CMB305 therapy.
- CMB305's safety profile consisted mostly of mild to moderate adverse events, with therapy being well tolerated by patients.
- Anti-NY-ESO-1 immune biomarkers identify cancer patients who may be more likely to have prolonged survival following therapy with CMB305
- Anti-NY-ESO-1 immune responses were observed in more than half of the patients who received CMB305 therapy.
- Induction of anti-NY-ESO-1 immunity in patients treated with CMB305 or LV305 was associated with better clinical outcomes, including survival.
- Immune biomarkers pre-treatment may guide regulatory strategy via the selection of patients more likely to respond to CMB305 therapy.
G100 Intratumoral Monotherapy with Radiation in Patients with Low-grade Follicular NHL (FL)
- More than 40% of the FL patients experienced objective responses based on WHO criteria (at least a 50% tumor reduction), including substantial tumor shrinkage in untreated, unirradiated distal (abscopal) lesions.
- Safety profile remains favorable at higher doses than those previously reported in Merkel cell carcinoma patients.
- G100 resulted in favorable tumor microenvironment changes.
1. An increased intratumoral expression of inflammatory cytokines/chemokines, T cell infiltration, and an increased frequency of clonal tumor infiltrating lymphocytes, were observed.
"The ability to identify patients who are likely to benefit from antigen-targeted immunotherapy has been an elusive goal. We believe the results highlighted here should be considered as we aim to maximize the chance of success of these novel modalities, including CMB305 and future product candidates from our ZVex platform." said Carlos Paya, M.D., Ph.D., President and Chief Executive Officer of Immune Design. "During the second half of the year, we hope to have the opportunity to build on these positive clinical and biomarker data for CMB305 and G100 monotherapy with the results from ongoing trials evaluating each agent in combination with anti-PD-1/PD-L1 inhibitors."
Presentations at American Society of Clinical Oncology Annual Meeting
Data underlying the topline releases above were published online today by the American Society of Clinical Oncology (ASCO) in abstracts accepted for presentation at ASCO's 2017 Annual Meeting in June (presentation information set forth below). The abstracts reflect an analysis performed on or before February 2017; additional data will be presented at the Annual Meeting.
ORAL PRESENTATION
Immune response, safety, and survival impact from CMB305 in NY-ESO-1+ recurrent soft tissue sarcomas (STS)
Abstract # 11006
Session Title: Sarcoma
Date: Friday, June 2, 2017
Time: 3 p.m. — 6 p.m. CT (oral session)
Location: S100bc
Presenter: Neeta Somaiah, M.D., Department of Sarcoma Medical Oncology, The University of Texas MD Anderson Cancer Center
POSTER PRESENTATIONS
The Association of CMB305 or LV305-induced and baseline anti-NY-ESO-1 immunity with survival in recurrent cancer patients
Abstract # 3090
Session Title: Developmental Therapeutics—Immunotherapy
Date: Monday, June 5, 2017
Time: 8 a.m. — 11:30 a.m. CT
Location: Hall A
Presenter: Seth M. Pollack, M.D., Fred Hutchinson Cancer Research Center
Intratumoral G100 to induce systemic immune responses and abscopal tumor regression in patients with follicular lymphoma
Abstract # 7537
Session Title: Hematologic Malignancies — Lymphoma and Chronic Lymphocytic Leukemia
Date: Monday, June 5, 2017
Time: 8 a.m. — 11:30 a.m. CT
Location: Hall A
Presenter: Christopher Flowers, M.D., Department of Hematology and Medical Oncology, Emory University School of Medicine
About CMB305
CMB305 is a prime-boost vaccine approach against NY-ESO-1-expressing tumors, designed to generate an integrated, anti-NY-ESO-1 immune response in vivo via a targeted, specific interaction with dendritic cells, a mechanism of action Immune Design believes differs from traditional cancer vaccines. CMB305 is being evaluated in STS patients in ongoing Phase 1 monotherapy and 2 combination studies with the anti-PD-L1 antibody, Tecentriq® (atezolizumab), pursuant to a collaboration with Genentech. Immune Design has received Orphan Drug Designation for CMB305 from the U.S. Food and Drug Administration (FDA) for the treatment of soft tissue sarcoma, as well as from the FDA and European Medicines Agency for each of the components of CMB305 for the treatment of soft tissue sarcoma.
About G100
G100 contains a potent synthetic small molecule toll-like receptor-4 (TLR-4) agonist, Glucopyranosyl Lipid A (GLA), and is the lead product candidate in Immune Design's Antigen Agnostic approach. It leverages the activation of both innate and adaptive immunity, including dendritic cells, in the tumor microenvironment to create an immune response against the tumor's preexisting diverse set of antigens. G100 is being evaluated as both a monotherapy (with XRT) and in combination with Merck's anti-PD-1 agent, Keytruda® (pembrolizumab), pursuant to a clinical collaboration with Merck, in a randomized Phase 1/2 trial in patients with follicular non-Hodgkin's lymphoma. The FDA has granted Orphan Drug Designation for G100 for the treatment of follicular non-Hodgkin's lymphoma.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the two leading product candidates focused in cancer immunotherapy, are the first products from its two separate discovery platforms targeting dendritic cells in vivo, ZVex® and GLAAS®. Both ZVex and GLAAS also have potential applications in infectious disease and allergy as demonstrated by ongoing pharmaceutical collaborations. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Cautionary Note on Forward-looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties that could cause our clinical development programs, future results or performance to differ significantly from those expressed or implied by the forward looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the progress, timing, scope and results of clinical trials, the association of data with treatment outcomes, and the timing and likelihood of obtaining regulatory approval of Immune Design's product candidates. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrolment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, the uncertainties and timing of the regulatory approval process, and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
Shari.Annes@immunedesign.com
650-888-0902
Series C growth round led by Glynn Capital Management with participation from GV
(formerly Google Ventures), along with existing venture and strategic investors
PLAYA VISTA, Calif. (April 25, 2017) – Science 37, a trailblazing company focused on “site-less” clinical trials, announced today $29 million in Series C funding led by Glynn Capital Management with participation from GV. The round included new strategic investments from Amgen Ventures, as well as participation from all existing investors Lux Capital, Redmile Group, dRx Capital (a Qualcomm and Novartis joint investment company), and Sanofi Ventures. Total invested capital since inception is now nearly $67 million.
“We’ve seen great success thus far in the model we developed. It has not only increased the number of people able to participate in clinical trials, but also reduced the time it takes to recruit by nearly 50 percent in some cases,” said Noah Craft, MD, PhD, and co-founder and CEO of Science 37. “The new funding will help us drive continued expansion and recruit top talent from both the technology and from the clinical trial industries. The round will also allow us to expand into new therapeutic areas and to introduce these trials to a much broader set of demographics, including those with limited access to major medical facilities where these trials are typically offered.”
Science 37’s core offering is focused on making clinical trials better for patients. The company is also directly working to address the lack of diversity represented in traditional trials. Currently only five percent of minorities are represented in all clinical trials and the percentage involved in cancer trials can be below two percent. Statistics such as these were a major driving force in Science 37’s focus on creating a metasite™ to carry out telemedicine-based clinical research. The metasite™ or “siteless” clinical trial model enables them to bridge the geographic gap that exists between many patients and research focused medical facilities.
Science 37 uses their proprietary metasite™ and technology platform, the Network Oriented Research Assistant (NORA®) to make clinical trials easier for patients to access from the comfort of their homes. The metasite™ decentralizes trials, enabling patients to participate regardless of their geographical location using their mobile devices and telemedicine services, and includes Virtual Trial Specialists, mobile nursing companies, and study monitors to execute end-to-end clinical trial services for sponsors.
“Science 37 has shown rapid growth since forming in September, 2014,” said Scott Jordon, Managing Director at Glynn Capital Management. “We believe the advances being made will fundamentally alter the landscape of research in the medical field and help drive more positive outcomes for all constituents.”
“Science 37 has developed a novel, decentralized framework for running clinical trials,” said Krishna Yeshwant, General Partner at GV. “The company has completed clinical trials with impressive early results, and has a great combination of operational expertise in clinical research, coupled with software sensibilities.”
The new funding will help speed additional development of the proprietary Science 37 NORA® software platform, to scale the clinical operations teams to meet the tremendous demand for upcoming trials, to help expand into new innovative markets and therapeutic areas, and to prepare for international expansion in the coming years.
“Many new drug candidates never get developed as a direct result of the long timelines and costs required to complete a traditional study,” said Belinda Tan, MD, PhD, and co-founder and chief medical officer of Science 37. “The long recruitment process for trials alone is one of the biggest drivers of these drug development costs. By streamlining that process we hope to help bring new cures to patients faster.”
About Science 37
Science 37 is a Los Angeles-based technology-enabled clinical research company. They created a new clinical trial operating model – the metasite™ – that unlocks access so researchers can find the right patients, and patients can find the right trials. Their Network Orientated Research Assistant, NORA®, is the cloud-based patient-centric mobile research platform that connects everyone – safely and securely. Science 37 offers end-to-end clinical trial services without geographic limitations, making clinical research faster to accelerate biomedical discovery. Ultimately, Science 37 enables better, faster people-powered science and brings clinical trials to scale. Learn more at http://science37.com, and follow Science 37 on Twitter @Science_37.
About Glynn Capital Management
Glynn Capital Management is focused on investments in leading private and public technology growth companies. We seek to be long-term investors in a limited number of excellent companies with world-class management teams, sustainable business models, and long-term growth potential. Glynn Capital has been investing in technology companies for several decades and is based in Menlo Park, California. More information is available at www.glynncapital.com or by emailing IR@glynncapital.com.
Media Contact:
Aunny Bathe
abathe@science37.com
310-367-9543
Pediatric Development is Planned Upon Completion of Preclinical Testing
NEW YORK, April 10, 2017 (GLOBE NEWSWIRE) -- Ovid Therapeutics, Inc., a privately held biopharmaceutical company committed to developing medicines that transform the lives of people with rare neurological diseases, today announced that it has initiated a Phase 1 clinical trial to evaluate the pharmacokinetics (PK), safety and tolerability of OV101 in adolescents diagnosed with Angelman syndrome or Fragile X syndrome. OV101 (gaboxadol), a delta (δ)-selective GABAA receptor agonist, is believed to be the first investigational drug to target the disruption of tonic inhibition, a key mechanism that allows a healthy human brain to decipher excitatory and inhibitory neurological signals correctly without being overloaded. Tonic inhibition is believed to play a significant role in the neurodevelopmental symptoms characteristic of disorders such as Angelman syndrome and Fragile X syndrome.
“Those with neurodevelopmental disorders such as Angelman syndrome and Fragile X syndrome are affected from birth. Our goal is to provide medicines to these individuals at all ages. The initiation of this trial represents the first step in our strategy to develop OV101 for an adolescent population. This is an important step because having access to medicines at a younger age has the potential to have a transformative effect on the lives of these patients and their families,” said Amit Rakhit, M.D., MBA, chief medical and portfolio officer of Ovid Therapeutics. “We look forward to continuing to partner with the Angelman and Fragile X syndrome communities as we work to develop therapies that can have the greatest impact on the challenges they face every day.”
The Phase 1 single dose, single-arm, open-label, clinical trial of OV101 will measure PK parameters. The trial is expected to enroll adolescent patients, ages 13 to 17, who have been diagnosed with either Angelman syndrome or Fragile X syndrome. Additional details on the Phase 1 clinical trial can be found at www.clinicaltrials.gov.
In addition to this clinical trial in adolescents, Ovid is planning to initiate clinical development in a pediatric population upon completing the required preclinical testing.
About Angelman Syndrome
Angelman syndrome is a rare genetic disorder that is characterized by a variety of signs and symptoms. Characteristic features of this disorder include delayed development, intellectual disability, severe speech impairment, problems with movement and balance, seizures, sleep disorders and anxiety. The most common cause of Angelman syndrome is the disruption of a gene that codes for ubiquitin protein ligase E3A (UBE3A). Angelman syndrome affects approximately 1 in 12,000 to 20,000 people in the United States. There are currently no United States Food and Drug Administration (FDA)-approved therapies for the treatment of Angelman syndrome.
Angelman syndrome is associated with a reduction in tonic inhibition, a function of the delta (δ)-selective GABAA receptor that allows a human brain to decipher excitatory and inhibitory neurological signals correctly without being overloaded. If tonic inhibition is reduced, the brain becomes inundated with signals and loses the ability to separate background noise from critical information.
About Fragile X Syndrome
Fragile X syndrome is the most common inherited form of intellectual disability and autism, with a prevalence of 1 in 3,600 to 4,000 males and 1 in 4,000 to 6,000 females in the United States. Individuals with Fragile X syndrome often have a range of behavioral challenges, such as cognitive impairment, anxiety, mood swings, hyperactivity, attention deficit, poor sleep, self-injury and heightened sensitivity to various stimuli, such as sound. Additionally, individuals with Fragile X syndrome are prone to comorbid medical issues including seizures and sleep disturbance. Fragile X syndrome results from mutations in the FMR1 gene, which blocks expression of the Fragile X Mental Retardation Protein (FMRP), an important protein in GABA synthesis. There are no FDA-approved therapies for Fragile X syndrome, and treatment primarily consists of behavioral interventions and pharmacologic management of symptoms.
In studies of individuals with Fragile X syndrome and in experimental models, extrasynaptic GABA levels are abnormally reduced, and there is also dysregulation of GABA receptors. This ultimately contributes to a decrease in tonic inhibition, causing the brain to become inundated with signals and lose the ability to separate background noise from critical information.
About OV101
OV101 (gaboxadol) is a delta (δ)-selective GABAA receptor agonist and is believed to be the first investigational drug to target the disruption of tonic inhibition. In preclinical models, OV101 has been able to selectively activate the δ-subunit of GABAA receptors, which are found in the extrasynaptic space (outside of the synapse), and help regulate neuronal activity through tonic inhibition.
Ovid is developing OV101 for use in both Angelman syndrome and Fragile X syndrome to potentially restore tonic inhibition and relieve several of the symptoms of these disorders. In preclinical studies, it was observed that OV101 improved symptoms of Angelman syndrome and Fragile X syndrome.
In September 2016, the FDA granted orphan drug designation for OV101 for the treatment of Angelman syndrome. The United States Patent and Trademark Office has granted Ovid two patents directed to methods of treating Angelman syndrome using THIP (OV101). The issued patents expire in 2035, without regulatory extensions.
About Ovid Therapeutics
Ovid Therapeutics is a privately held, New York-based, biopharmaceutical company using its BoldMedicineTM approach to develop therapies that transform the lives of patients with rare neurological diseases. Ovid’s drug candidate, OV101, is currently in development for the treatment of symptoms of Angelman syndrome and Fragile X syndrome. Ovid has initiated the Phase 2 STARS trial of OV101 in adults with Angelman syndrome and a Phase 1 trial in adolescents with Angelman syndrome or Fragile X syndrome. Ovid is also developing OV935 in collaboration with Takeda Pharmaceutical Company Limited for the treatment of rare epileptic encephalopathies. OV935 is expected to initiate a Phase 1b/2a trial in rare epileptic encephalopathies in 2017.
For more information on Ovid, please visit http://www.ovidrx.com/.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements. Forward-looking statements contained in this press release include, without limitation, statements regarding Ovid’s expectations regarding the clinical development of OV101 for the treatment of Angelman syndrome or Fragile X syndrome, the clinical benefits of OV101 for people with Angelman syndrome or Fragile X syndrome, the expected enrollment in the Phase 1 trial and the timing of clinical trials. Words such as “may,” “believe,” “will,” “expect,” “plan,” “anticipate,” “estimate,” “intend” and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are not guarantees of future performance and involve a number of unknown risks, assumptions, uncertainties and factors that are beyond Ovid’s control. All forward-looking statements are based on Ovid’s expectations and assumptions as of the date of this press release. Actual results may differ materially from these forward-looking statements. Except as required by law, Ovid expressly disclaims any responsibility to update any forward-looking statement contained herein, whether as a result of new information, future events or otherwise.
Contacts
Investors:
Burns McClellan
Steve Klass, 212-213-0006
Sklass@burnsmc.com
Media:
Pure Communications, Inc.
Katie Engleman, 910-509-3977
katie@purecommunicationsinc.com

ROCKVILLE, Md.--(BUSINESS WIRE)-- GlycoMimetics, Inc. (NASDAQ:GLYC) today announced that the first of two patient cohorts in its Phase 2 acute myeloid leukemia (AML) trial of GMI-1271 has completed enrollment. This cohort is comprised of 25 patients 60 years of age or older with newly diagnosed AML. The study is designed to evaluate the potential of GMI- 1271, GlycoMimetics' E-selectin antagonist drug candidate, in combination with chemotherapy, as a treatment for patients with both newly diagnosed and relapsed/refractory AML. Enrollment in the study's second arm is expected to complete in the middle of this year. The two arms combined will enroll a total of about 90 patients.
"At the ASH meeting in December, we showed that GMI-1271 was well-tolerated and demonstrated a high remission rate among the patient volunteers who were enrolled early in the study," said Helen Thackray, M.D., Chief Medical Officer of GlycoMimetics. "We are enthusiastic about that data, and as such, we look forward to opportunities later in the year to report initial treatment outcomes for this study."
GMI-1271 data were presented in 2016 at meetings of the European Hematology Association (EHA) and the American Society of Hematology (ASH), showing high remission rates and lower than expected 30- and 60-day mortality rates in early evaluations of patients with relapsed/refractory AML.
In addition to the ongoing Phase 1/2 trial, clinical investigators are currently evaluating GMI-1271 in an ongoing Phase 1 clinical trial in multiple myeloma. Preclinical data supporting the multiple myeloma study is scheduled to be shared in an oral presentation at the American Association of Cancer Research (AACR) Annual Meeting 2017 in April. Specifically, the newly announced preclinical results show a strong effect on cancer cells in combination with chemotherapy and are supportive of the ongoing Phase 1 clinical studies of GMI-1271 in multiple myeloma.
About AML
Acute myeloid leukemia (AML) is a cancer of the blood and bone marrow. AML is the most common type of acute leukemia in adults. Each year in the United States, about 19,900 people (usually older than 45 years of age) are diagnosed, and about 10,400 people die from all forms of the disease, according to the American Cancer Society. Unlike other cancers that start in an organ and spread to the bone marrow, AML is known for rapid growth of abnormal white blood cells that gather in the bone marrow, getting in the way of normal blood cell production. The lack of normal blood cells can cause some of the symptoms of AML, including anemia (shortage of red blood cells resulting in tiredness and weakness), neutropenia (shortage of white blood cells that may lead to increased infections), and thrombocytopenia (shortage of platelets in the blood that may lead to excessive bleeding). Current treatment options for AML consist of reducing and eliminating cancer cells mainly through chemotherapy, radiation therapy, and stem cell transplantation.
About GlycoMimetics, Inc.
GlycoMimetics is a clinical-stage biotechnology company focused on cancer and sickle cell disease. GlycoMimetics' most advanced drug candidate, rivipansel, a pan-selectin antagonist, is being developed for the treatment of vaso-occlusive crisis in sickle cell disease and is being evaluated in a Phase 3 clinical trial being conducted by its strategic collaborator, Pfizer. GlycoMimetics' wholly-owned drug candidate, GMI-1271, an E-selectin antagonist, is being evaluated in an ongoing Phase 1/2 clinical trial as a potential treatment for AML and in a Phase 1 clinical trial in multiple myeloma. GlycoMimetics has also recently initiated a clinical trial with a third drug candidate, GMI-1359, a combined CXCR4 and E-selectin antagonist. GlycoMimetics is located in Rockville, MD in the BioHealth Capital Region. Learn more at www.glycomimetics.com.
Forward-Looking Statements
This press release contains forward-looking statements regarding GlycoMimetics' planned activities with respect to the clinical development of its drug candidate, GMI-1271. Actual results may differ materially from those indicated by such forward-looking statements as a result of various important factors, including the availability and timing of data from ongoing clinical trials, the uncertainties inherent in the initiation of future clinical trials, whether interim results from a clinical trial will be predictive of the final results of the trial or results of early clinical trials will be indicative of the results of future trials, expectations for regulatory approvals, availability of funding sufficient for GlycoMimetics' foreseeable and unforeseeable operating expenses and capital expenditure requirements, other matters that could affect the availability or commercial potential of GlycoMimetics' drug candidates and other factors discussed in the "Risk Factors" section of GlycoMimetics' Annual Report on Form 10-K that was filed with the U.S. Securities and Exchange Commission on February 29, 2016, and other filings GlycoMimetics makes with the Securities and Exchange Commission from time to time. In addition, the forwardlooking statements included in this press release represent GlycoMimetics' views as of the date hereof. GlycoMimetics anticipates that subsequent events and developments may cause its views to change. However, while GlycoMimetics may elect to update these forward-looking statements at some point in the future, GlycoMimetics specifically disclaims any obligation to do so, except as may be required by law. These forward-looking statements should not be relied upon as representing GlycoMimetics' views as of any date subsequent to the date hereof.
View source version on businesswire.com: http://www.businesswire.com/news/home/20170307005569/en/
For GlycoMimetics, Inc.
Investors:
Shari Annes, 650-888-0902
sannes@annesassociates.com
or
Media:
Jamie Lacey-Moreira, 410-299-3310
jamielacey@presscommpr.com
Source: GlycoMimetics, Inc.
News Provided by Acquire Media
Preclinical Data Demonstrates Tumor Eradication with "Prime-Pull" Immunotherapy Approach Combining a ZVex® vector and G100
— ZVex Causes Potent Activation of Dendritic Cells —
SEATTLE and SOUTH SAN FRANCISCO, Calif., March 01, 2017 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on oncology, today announced that new preclinical data showing the broad anti-tumor activity on its "prime-pull" approach, as well as the ability of its ZVex vectors to activate dendritic cells potently, will be presented at the upcoming American Association for Cancer Research (AACR) 2017 Annual Meeting, being held from April 1-5, 2017 in Washington D.C.
"These AACR data illustrate the significant local and systemic immune responses that the combination of ZVex vectors with G100 may generate. We observed complete eradication of large, established B16 tumors in animal models, which previously has been achieved only with complex regimens," said Jan ter Meulen, MD, PhD, Chief Scientific Officer at Immune Design. "In addition, we will present additional data supporting the ability of ZVex vectors to generate potent activation of human dendritic cells."
Immune Design will present data showing the "Prime-Pull" concept that involves the (i) intradermal administration of the dendritic cell (DC)-targeting ZVex vector expressing a tumor-associated antigen and (ii) intratumoral injection of G100, a formulated, potent synthetic toll-like receptor-4 (TLR-4) agonist, in the difficult to treat B16 melanoma model. Antigenspecific CD8 T-cells are induced ("primed") by a ZVex vector, and subsequent injection of G100 leads to pro-inflammatory changes in the tumor microenvironment (TME), which induces the trafficking of T-cells into the tumor (the "pull"). This inflamed TME and recruitment of ZVex-induced CD8 T cells eradicated large, established B16 tumors. Also importantly, treated mice rejected re-challenge with a tumor lacking the antigen used for immunization, indicating antigen spreading induced by the immunotherapeutic regimen. Immune Design is evaluating this immunotherapy approach in an ongoing Phase 1 trial in patients with soft tissue sarcoma who are receiving G100 and CMB305, its prime-boost approach that is being evaluated in multiple clinical trials as both a monotherapy and in combination with atezolizumab, Genentech's anti-PDL1 antibody.
In addition, Immune Design will present separate data highlighting the ability of its ZVex vectors to induce potent, innate immune activation in human DCs. Company researchers studied the effect of human DC transduction with ZVex vectors by gene expression profiling. Human DCs transduced with ZVex vectors displayed statistically significant up-regulation of genes involved in antigen presentation and anti-viral defense pathways, highlighting that ZVex is sufficient to activate transduced DCs and facilitate antigen presentation to T cells.
The details for the poster presentations are as follows:
Large established B16 tumors in mice are eradicated by ZVex® (dendritic cell-targeting lentiviral vector) and G100 (TLR4 agonist) combination immunotherapy through increasing tumor-infiltrating effector T cells and inducing antigen spreading
Abstract #: 5673
Session Category: Clinical Research
Session Title: Innate Immunity to Generate Adaptive Immunity
Date and Time: Wednesday, April 5, 2017, 8:00 a.m. — 12:00 p.m.
Location: Convention Center, Halls A-C, Poster Section 28
Poster Board: 27
Authors: Tina C. Albershardt, Andrea J. Parsons, Jardin Leleux, Peter Berglund, Jan ter Meulen. Immune Design
The poster presentation will be made available at Immune Design's website on or after April 5, 2017.
ZVex® lentiviral vector strongly activates pro-inflammatory, antigen processing, and anti-viral defense response pathways in monocyte-derived dendritic cells
Abstract #: 5092
Session Category: Experimental and Molecular Therapeutics
Session Title: Gene- and Vector-based Therapy
Date and Time: Wednesday, April 5, 2017, 8 a.m. — 12 p.m.
Location: Convention Center, Halls A-C, Poster Section 3
Poster Board: 8
Authors: Anshika Bajaj, Lisa Y. Ngo, Peter Berglund, Jan ter Meulen. Immune Design
The poster presentation will be made available at Immune Design's website on or after April 5, 2017.
About ZVex
ZVex is Immune Design's discovery platform designed to activate and expand the immune system's natural ability to create tumor-specific cytotoxic T cells (CTLs) in vivo. ZVex uses a re-engineered virus to carry genetic information of a tumor antigen selectively to dendritic cells in the skin or lymph nodes. This ultimately results in the creation of CTLs designed to kill tumor cells bearing that same specific tumor antigen. ZVex is also designed to carry the genetic information for, and therefore potentially cause dendritic cells to express, multiple antigens and/or selected epitopes of interest (including neoantigens), as well as cytokines or other immunomodulatory molecules.
About G100
G100 is a product candidate from Immune Design's GLAAS® discovery platform. It contains a potent synthetic small molecule toll-like receptor-4 (TLR-4) agonist, Glucopyranosyl Lipid A (GLA), and is the lead product candidate in Immune Design's Antigen Agnostic approach. It leverages the activation of both innate and adaptive immunity, including Dendritic Cells, in the tumor microenvironment to create an immune response against the tumor's preexisting diverse set of antigens. A growing set of clinical and preclinical data have demonstrated the ability of G100 to activate tumor-infiltrating lymphocytes, macrophages and dendritic cells, and promote antigen-presentation and the recruitment of T cells to the tumor. The ensuing induction of local and systemic immune responses has been shown to result in local and abscopal (shrinking of tumors outside the scope of the localized treatment) tumor control in preclinical studies. G100 was evaluated in a Phase 1 study in Merkel cell carcinoma patients and produced a 50% overall response rate per protocol and a favorable safety profile. Currently, G100 is being evaluated as both a monotherapy with local radiation and in combination with Merck's anti- PD-1 agent, pembrolizumab, pursuant to a clinical collaboration with Merck, in a randomized Phase 1/2 trial in patients with follicular non-Hodgkin lymphoma.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the primary foci of Immune Design's ongoing immunooncology clinical programs, are products of its two synergistic discovery platforms, ZVex and GLAAS, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the timing of initiation, progress, scope and outcome of clinical trials for Immune Design's product candidates and the reporting of clinical data regarding Immune Design's product candidates. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment and unexpected litigation or other disputes. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
Without clinical trials, new medicine may never make it from the research lab to patients in need. These carefully designed studies can provide important data that include proper dosage, benefit to patients, and potential side effects.
There is a growing challenge, however, in finding appropriate participants, especially for treatments that target highly specific conditions affecting narrower patient populations. Right now, there are more than 40,000 clinical studies recruiting patients in the U.S. alone, with some requiring thousands of participants, each of whom must meet precise criteria to join. So it’s not surprising that 80% of these important studies are delayed due to recruitment problems, according to a study by the Center for Information and Study on Clinical Research Participation (CISCRP).
Unfortunately, those delays mean it can take longer for innovative new medicines to be studied and approved, leaving patients to wait years for new treatment options. To tackle this growing problem, Sanofi is taking a digital approach to clinical trials, partnering with Science 37, a clinical research services and technology company based in California.
Leveraging mobile technology and telemedicine capabilities, this new approach will allow Sanofi to develop “site-less” or decentralized clinical trials that are more patient friendly: easier for them to access, and eliminating many of the common impediments to participation. Using digital technologies to streamline finding and retaining participants for the entire length of a study has the potential to reduce the time required for a typical trial by at least 30%, according to Science 37.
“After years invested in the lab on an innovative treatment, the clinical trials are where we finally obtain and analyze the relevant data that will let us understand how well a new treatment will benefit patients,” said Lionel Bascles, Global Head of Clinical Sciences and Operations of Sanofi. “With digital clinical trials we can get and analyze the data on how a new medicine works in the real world a lot sooner, which means patients get the medicines they need sooner.”
Going digital also eliminates a number of other hurdles to patient participation, including the most significant: geography.
Most people are eager to participate in relevant trials – 87% of patients want to do so, the CISCRP study found. Yet, 70% of potential participants live more than two hours away from the nearest study center. Because most clinical trials require patients to travel to those centers for regular tests and observations, sometimes several times each week for the duration of the trial, this distance is another challenge to patient access.
Science 37’s approach allows patients to be monitored and report to researchers via an Apple iPhone equipped with the company’s NORA® technology. Qualified study participants are provided with the phone, a data plan and any other sensors or connected devices needed for the trial, along with the medicines being researched. Participants can reach study staff at any time via the mobile device, while also remaining under the care of their local health care professionals. Mobile nurses are also sent to the participant’s home to provide services like blood draws when needed, and nearby hospitals or clinics are engaged for scans or other tests that require specialized equipment.
The patient’s data are sent securely to researchers who can immediately access information that would otherwise have to be collected by medical personnel through face-to-face interactions at study centers. This platform can also remind patients to take their study medications at the proper time, and let researchers know if participants are adhering to the study requirements.
“Our decentralized clinical trial model addresses critical shortcomings of traditional clinical trials, such as enrolling and retaining appropriate patients. Whether you live near a major research institution, or in a remote area, we make participation possible,” said Noah Craft, CEO of Science 37. “By utilizing a patient’s home in lieu of a physical trial site, we remove the burden of travel for those too sick or remote and provide access to qualified individuals who want to volunteer for a study but cannot because of geographic limitations.”
The Science 37 platform will also help engage patients who would normally not participate in clinical trials, “so our data will much more closely track the diversity of the population,” Bascles said. “In addition to reducing the burden for patients, decentralized clinical trials are far more likely to keep patients engaged for the full length of the trial, increasing the relevance and the acceptability of the data by regulators.”
Sanofi’s agreement with Science 37 covers use of its Metasite™ model and NORA technology across the U.S. with plans to expand internationally in the future. By eliminating months of searching for patients and long travel time to study sites, virtual clinical trials could reduce total trial time by as much as two years.
Partnering with Science 37 is the most recent strengthening of the relationship with Sanofi, which began last October when Sanofi’s venture capital fund, Sanofi-Genzyme BioVentures, made a minority investment in Science 37.
“Science 37 has a great track record, and they are smart and forward-thinking about developing the science around clinical trials that leverage digital technologies,” said Heather Bell, Global Head of Digital and Analytics for Sanofi. “As part of the scope of our digital strategy, we have expanded the scope of the venture fund to include digital investments, and Science 37 was our first investment since that change and we’re very excited about it.”
SEATTLE and SOUTH SAN FRANCISCO, Calif., Feb. 22, 2017 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on oncology, today announced that the U.S. Food and Drug Administration (FDA) has granted Orphan Drug Designation for its investigational intratumoral therapy, G100, for the treatment of follicular non-Hodgkin's lymphoma.
Orphan Drug Designation is granted by the FDA Office of Orphan Drug Products to products that treat rare diseases. The FDA defines rare diseases as those affecting fewer than 200,000 people in the United States. Orphan Drug Designation provides the sponsor certain benefits and incentives, including a period of marketing exclusivity for the first marketing application, if regulatory approval is received for the designated indication, potential tax credits for certain activities and waiver of certain administrative fees.
G100 is a product candidate from Immune Design's GLAAS® discovery platform. It contains a potent synthetic small molecule toll-like receptor-4 (TLR-4) agonist, Glucopyranosyl Lipid A (GLA), and is the lead product candidate in Immune Design's Antigen Agnostic approach. It leverages the activation of both innate and adaptive immunity, including Dendritic Cells, in the tumor microenvironment to create an immune response against the tumor's preexisting diverse set of antigens. A growing set of clinical and preclinical data have demonstrated the ability of G100 to activate tumor-infiltrating lymphocytes, macrophages and dendritic cells, and promote antigen-presentation and the recruitment of T cells to the tumor. The ensuing induction of local and systemic immune responses has been shown to result in local and abscopal (shrinking of tumors outside the scope of the localized treatment) tumor control in preclinical studies. G100 was evaluated in a Phase 1 study in Merkel cell carcinoma patients and produced a 50% overall response rate per protocol and a favorable safety profile. Currently, G100 is being evaluated as both a monotherapy (with local radiation) and in combination with Merck's anti-PD-1 agent, pembrolizumab, pursuant to a clinical collaboration with Merck, in a randomized Phase 1/2 trial in patients with follicular non-Hodgkin's lymphoma.
CMB305, Immune Design's prime-boost cancer immunotherapy product candidate, has previously been designated orphan drug status for soft tissue sarcoma by the FDA, and each of the two agents comprising CMB305 also have both U.S. and European Orphan drug designation for soft tissue sarcoma.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the primary foci of Immune Design's ongoing immunooncology clinical programs, are products of its two synergistic discovery platforms, ZVex® and GLAAS, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the progress, scope and outcome of clinical trials for Immune Design's product candidates, the reporting of clinical data regarding Immune Design's product candidates and the timing and likelihood of regulatory filings and approvals. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, and unexpected litigation or other disputes. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful. Orphan Drug Designation does not provide any assurance of regulatory approval or expedite regulatory review. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
STARS is the first industry-sponsored clinical trial for Angelman syndrome
NEW YORK, Feb. 09, 2017 (GLOBE NEWSWIRE) -- Ovid Therapeutics, a privately held biopharmaceutical company committed to developing medicines that transform the lives of people with rare neurological disorders, today announced it has randomized the first patient in its STARS trial – a Phase 2 clinical trial of OV101 in adults with Angelman syndrome. OV101 (gaboxadol), a delta (δ)-selective GABAA receptor agonist, is believed to be the first investigational drug to target the disruption of tonic inhibition, a key mechanism that allows a healthy human brain to decipher excitatory and inhibitory neurological signals correctly without being overloaded. Tonic inhibition is believed to play a significant role in the developmental and neurological symptoms characteristic of disorders such as Angelman syndrome and Fragile X syndrome.
“Angelman syndrome is a rare genetic disorder associated with a broad spectrum of symptoms such as development delay, seizures, balance problems and sleep disturbance, resulting in the need for life-long care,” said Lynne Bird, M.D., principal investigator and professor of clinical pediatrics at the University of California San Diego and Rady Children's Hospital-San Diego. “There are currently no FDA approved therapies for the treatment of Angelman Syndrome. The standard of care provides limited treatment options for Angelman syndrome, most of which are general supportive therapies that are used for many other disorders and don’t address the specific pathophysiology of Angelman syndrome. I believe the STARS trial will be the first trial to study an investigative drug that may have the opportunity to address several symptoms that characterize the disorder and that OV101 has the potential to offer a meaningful clinical benefit for people with Angelman syndrome.”
“We are proud to have partnered with the Angelman syndrome community in the design of the STARS trial and thrilled that we have taken an important step in advancing the development of a potential treatment for Angelman syndrome that may benefit the families and individuals living with this disorder,” said Amit Rakhit, M.D., MBA, chief medical and portfolio management officer of Ovid Therapeutics. “OV101 highlights our commitment to developing transformative therapies for people and their families living with rare disorders of the brain.”
About the Phase 2 STARS Trial
The STARS trial is a randomized, double-blind, placebo-controlled Phase 2 clinical trial investigating the safety and efficacy of OV101 that was designed in consultation with the Angelman syndrome community. The trial is expected to enroll approximately 75 adults in the United States aged 18-49 years with a confirmed diagnosis of Angelman syndrome. The primary endpoint of the trial is to assess the safety and tolerability of OV101. Additionally, the trial has several exploratory endpoints to evaluate measures of gross and fine motor skills, maladaptive behavior, sleep, clinical global impression and health-related quality of life questionnaires.
Learn more about the STARS trial at www.clinicaltrials.gov.
About Angelman Syndrome
Angelman syndrome is a rare genetic disorder that is characterized by a variety of signs and symptoms. Characteristic features of this disorder include delayed development, intellectual disability, severe speech impairment, problems with movement and balance, seizures, sleep disorders and anxiety. The most common cause of Angelman syndrome is the disruption of a gene that codes for ubiquitin protein ligase E3A (UBE3A). Angelman syndrome affects approximately 1 in 12,000 to 20,000 people in the United States. There are currently no FDA approved therapies for the treatment of Angelman syndrome.
Angelman syndrome is associated with a reduction in tonic inhibition, a function of the delta (δ)-selective GABAA receptor that allows a human brain to decipher excitatory and inhibitory neurological signals correctly without being overloaded. If tonic inhibition is reduced, the brain becomes inundated with signals and loses the ability to separate background noise from critical information.
About OV101
OV101 (gaboxadol) is a delta (δ)-selective GABAA receptor agonist and is believed to be the first investigational drug to target the disruption of tonic inhibition, a key mechanism that allows a healthy human brain to decipher excitatory and inhibitory neurological signals correctly without being overloaded. Loss of tonic inhibition is implicated in a host of rare neurological disorders and is established in genetic models. In preclinical models, OV101 has been able to selectively activate the δ-subunit of GABAA receptors, which are found in the extrasynaptic space (outside of the synapse), and helped regulate neuronal activity through tonic inhibition.
Ovid is developing OV101 for use in both Angelman syndrome and Fragile X syndrome to potentially restore tonic inhibition and relieve several of the symptoms of these disorders. In preclinical studies, it was observed that OV101 improved symptoms of Angelman syndrome and Fragile X syndrome.
In September 2016, the United States Food and Drug Administration granted orphan drug designation for OV101 for the treatment of Angelman syndrome. The United States Patent and Trademark Office has granted Ovid two patents directed to methods of treating Angelman syndrome using THIP (OV101). The issued patents expire in 2035, without regulatory extensions.
About Ovid Therapeutics
Ovid Therapeutics is a privately held, New York-based, biopharmaceutical company using its BoldMedicineTM approach to develop therapies that transform the lives of patients with rare neurological disorders. Ovid’s drug candidate, OV101, is currently in development for the treatment of symptoms of Angelman syndrome and Fragile X syndrome. Ovid is also developing OV935 in collaboration with Takeda Pharmaceutical Company Limited for the treatment of rare epileptic encephalopathies. Ovid has initiated a Phase 2 STARS trial of OV101 in adults with Angelman syndrome and Ovid intends to commence a Phase 1 trial in adolescents with Angelman syndrome or Fragile X syndrome. OV935 is expected to commence a Phase 1b/2a trial in rare epileptic encephalopathies in 2017.
For more information, visit http://www.ovidrx.com.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements. Forward-looking statements contained in this press release include, without limitation, statements regarding Ovid’s expectations regarding the clinical development of OV101, the clinical benefits of OV101 for people with Angelman syndrome and the expected enrollment in the STARS trial. Words such as “may,” “believe,” “will,” “expect,” “plan,” “anticipate,” “estimate,” “intend” and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are not guarantees of future performance and involve a number of unknown risks, assumptions, uncertainties and factors that are beyond Ovid’s control. All forward-looking statements are based on Ovid’s expectations and assumptions as of the date of this press release. Actual results may differ materially from these forward-looking statements. Except as required by law, Ovid expressly disclaims any responsibility to update any forward-looking statement contained herein, whether as a result of new information, future events or otherwise.
Contacts
Investors:
Burns McClellan
Steve Klass, 212-213-0006
Sklass@burnsmc.com
Media:
Pure Communications, Inc.
Katie Engleman, 910-509-3977
katie@purecommunicationsinc.com
– Partnership Marks First Step in Ovid Therapeutics’ Commitment to Biomarker Development –
NEW YORK and MADISON, Wis., Feb. 09, 2017 (GLOBE NEWSWIRE) -- Ovid Therapeutics, a privately held biopharmaceutical company committed to developing medicines that transform the lives of people with rare neurological disorders, and NeuroPointDX, a privately held biotech company based in Madison, Wisconsin and Cambridge, Massachusetts, today announced that they have entered into a collaboration to identify novel biomarkers of Angelman syndrome by analyzing metabolomic profile data as part of Ovid’s ongoing randomized, double-blind, placebo-controlled Phase 2 clinical trial (STARS).
Angelman syndrome is a rare genetic disorder that is characterized by a variety of signs and symptoms, including delayed development, intellectual disability, severe speech impairment, problems with movement and balance, seizures, sleep disorders and anxiety. Like many other neurological disorders, symptoms and response to treatments vary widely from person to person.
“With the initiation of the STARS trial and this collaboration, we are making important progress in better understanding Angelman syndrome and developing much needed treatment options. The metabolic profile of this syndrome is not well understood, and we believe this biomarker study will produce critical data to fill this gap and inform us about the impact of OV101 as a potential treatment option,” said Matthew During, M.D., DSc, FACP, FRACP, president and chief scientific officer of Ovid. “This collaboration is the first step in Ovid’s broader rare neurological disorder biomarker strategy to identify molecular markers of treatment responders and guide enrollment of participants in our clinical trials.”
“We are excited to partner with Ovid Therapeutics on this biomarker collaboration, particularly as it furthers our mission to improve the lives of people impacted by neurological disorders by identifying biomarkers that can improve diagnosis and inform more precise treatment strategies,” said Elizabeth Donley, chief executive officer of NeuroPointDX.
NeuroPointDX uses its metabolomics platform technology to identify differences in children with autism spectrum disorders compared to typically-developing children and between subgroups of children on the spectrum for earlier diagnosis and more precise treatment. The collaboration will leverage NeuroPointDX’s expertise in metabolomics in an effort to identify biomarkers associated with Angelman syndrome. Metabolomics is a process that allows comprehensive exploration of changes in small molecules present in the metabolism of patients to provide insight into the physiology of a disease and the response to treatment. In the STARS trial, this analysis is designed to provide molecular insights into disease mechanism and assess the potential response to OV101 to help understand the physiological impact of the compound in people with Angelman syndrome. This study may help identify the individuals that are most likely to respond to treatment.
About Angelman Syndrome
Angelman syndrome is a rare genetic disorder that is characterized by a variety of signs and symptoms. Characteristic features of this disorder include delayed development, intellectual disability, severe speech impairment, problems with movement and balance, seizures, sleep disorders and anxiety. The most common cause of Angelman syndrome is the disruption of a gene that codes for ubiquitin protein ligase E3A (UBE3A). Angelman syndrome affects approximately 1 in 12,000 to 20,000 people in the United States. There are currently no FDA-approved therapies for the treatment of Angelman syndrome.
Angelman syndrome is associated with a reduction in tonic inhibition, a function of the delta (δ)-selective GABAA receptor that allows a human brain to decipher excitatory and inhibitory neurological signals correctly without being overloaded. If tonic inhibition is reduced, the brain becomes inundated with signals and loses the ability to separate background noise from critical information.
About OV101
OV101 (gaboxadol) is a delta (d)-selective GABAA receptor agonist and is believed to be the first investigational drug to target the disruption of tonic inhibition, a key mechanism that allows a healthy human brain to decipher excitatory and inhibitory neurological signals correctly without being overloaded. Loss of tonic inhibition is implicated in a host of rare neurological disorders and is established in genetic models. In preclinical models, OV101 has been able to selectively activate the δ-subunit of GABAA receptors, which are found in the extrasynaptic space (outside of the synapse), and helped regulate neuronal activity through tonic inhibition.
Ovid is developing OV101 for use in both Angelman syndrome and Fragile X syndrome to potentially restore tonic inhibition and relieve several of the symptoms of these disorders. In preclinical studies, it was observed that OV101 improved symptoms of Angelman syndrome and Fragile X syndrome.
In September 2016, the United States Food and Drug Administration granted orphan drug designation for OV101 for the treatment of Angelman syndrome. The United States Patent and Trademark Office has granted Ovid two patents directed to methods of treating Angelman syndrome using THIP (OV101). The issued patents expire in 2035, without regulatory extensions.
About Ovid Therapeutics
Ovid Therapeutics is a privately held, New York-based, biopharmaceutical company using its BoldMedicineTM approach to develop therapies that transform the lives of patients with rare neurological disorders. Ovid’s drug candidate, OV101, is currently in development for the treatment of symptoms of Angelman syndrome and Fragile X syndrome. Ovid is also developing OV935 in collaboration with Takeda Pharmaceutical Company Limited for the treatment of rare epileptic encephalopathies. Ovid has initiated a Phase 2 STARS trial of OV101 in adults with Angelman syndrome and Ovid intends to commence a Phase 1 trial in adolescents with Angelman syndrome or Fragile X syndrome. OV935 is expected to commence a Phase 1b/2a trial in rare epileptic encephalopathies in 2017.
For more information, visit http://www.ovidrx.com/.
About NeuroPointDX
NeuroPoinDX is the diagnostics division of Stemina Biomarker Discovery. Stemina is a biotechnology company based in Madison, WI and Cambridge, MA that has developed a robust and reproducible proprietary platform for identifying changes in metabolism (“biomarkers”) utilizing highly sensitive analytical equipment and its proprietary platform technology. The Company’s diagnostics division, NeuroPointDX, identifies biomarkers of neurological disorders in cellular models of disease and human samples. These biomarkers provide greater insight into the pathways that are disrupted in patients with neurological disorders. This information can be translated into clinical diagnostics, individualized treatment recommendations, and new potential therapies.
The first area in which the Company is conducting clinical studies is in patients with autism and other neuro-developmental disorders. In September 2015, Stemina launched its 1,500 patient study the Children’s Autism Metabolome Project (CAMP). The study is funded by a $2.7 million grant from the National Institutes of Mental Health and an investment by the Nancy Lurie Marks Family Foundation. The CAMP study is the most comprehensive study of the metabolism of children with autism, and other neuro-developmental disorders ever conducted. This study may result in a panel of validated blood tests for autism that will more accurately diagnose and inform individualized treatment decisions based on the child’s own metabolism.
For more information about NeuroPointDX and Stemina, visit www.neuropointdx.com and www.stemina.com.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements. Forward-looking statements contained in this press release include, without limitation, statements regarding the collaboration with NeuroPointDX to identify novel biomarkers of Angelman syndrome and Ovid’s progress and expectations regarding the clinical development of OV101. Words such as “may,” “believe,” “will,” “expect,” “plan,” “anticipate,” “estimate,” “intend” and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are not guarantees of future performance and involve a number of unknown risks, assumptions, uncertainties and factors that are beyond Ovid’s control. All forward-looking statements are based on Ovid’s expectations and assumptions as of the date of this press release. Actual results may differ materially from these forward-looking statements. Except as required by law, Ovid expressly disclaims any responsibility to update any forward-looking statement contained herein, whether as a result of new information, future events or otherwise.
Ovid Contacts:
Investors:
Burns McClellan
Steve Klass, 212-213-0006
Sklass@burnsmc.com
Media:
Pure Communications, Inc.
Katie Engleman, 910-509-3977
katie@purecommunicationsinc.com
NeuroPointDX Contact:
Elizabeth Donley
Chief Executive Officer
608-577-9209
bdonley@neuropointdx.com
Renowned Pioneer to Help Shape the Company’s Growing Focus on Orphan Epilepsies
NEW YORK—January 19, 2017—Ovid Therapeutics, a privately held biopharmaceutical company committed to developing medicines that transform the lives of people with rare neurological diseases, today announced the appointment of Jacqueline A. French, M.D., a world-renowned expert in the treatment of epilepsy, to its Scientific Advisory Board.
“With our recent collaboration with Takeda Pharmaceutical Company Limited (TSE: 4502) to develop OV935 for orphan epileptic encephalopathies along with our preclinical research programs, we have established a solid foundation in developing treatments for rare disorders of epilepsy. The appointment of Dr. French to our Scientific Advisory Board furthers our commitment to this area as a core focus of the company with the goal to develop transformational medicines for patients and their families,” said Matthew During, M.D., DSc, FACP, FRACP, president and chief scientific officer of Ovid. “Dr. French has been deeply involved in the development of many of the key drugs in this area. In addition, she has spearheaded recent discussion with the FDA and EMA to create new clinical trial designs in epilepsy. With her experience as a preeminent clinical and research-focused epileptologist, we believe she will make a significant contribution to our thinking and approach. We are proud to have her as a member of our Scientific Advisory Board.”
“New understanding of the causes of epilepsy are emerging, giving rise to the opportunity to develop new therapies. This is particularly critical for many of the less common forms of epilepsy where few treatment options exist,” said Dr. French. “With Ovid’s strong neurology capabilities, interesting clinical and preclinical assets, and commitment to these rare but important disorders, I am excited to partner with their team so together, we can take a meaningful step in the development of treatments with the potential to have a significant impact on the lives of people with epilepsy.”
Dr. French is a professor in the Department of Neurology, director of Translational Research & Clinical Trials in Epilepsy, and a neurologist at the Comprehensive Epilepsy Center at NYU Langone Medical Center. She is also a founder and director of the Epilepsy Study Consortium, an academic group that has performed a number of early-phase clinical trials in epilepsy. She chaired an American Academy of Neurology (AAN)/American Epilepsy Society (AES) committee that developed two guidelines on the use of new antiepileptic drugs and has helped create guidelines for the International League Against Epilepsy. She currently serves as chief scientific officer of the Epilepsy Foundation. Previously, she was president and served on the board of the AES and was the secretary of the American Society of Experimental Neurotherapeutics. She has authored more than 200 articles and chapters, is the editor of three books and lectures internationally on clinical trials and the use of antiepileptic drugs. She was the 2005 recipient of the AES Service Award and the 2013 Epilepsy Foundation Hero award. Dr. French received a medical degree from Brown University. She completed her residency in neurology at Mount Sinai Hospital in New York and completed fellowships in epilepsy at Mount Sinai Hospital and Yale University.
About Ovid Therapeutics
Ovid Therapeutics is a privately held, New York-based, biopharmaceutical company using its BoldMedicineTM approach to develop therapies that transform the lives of patients with rare neurological disorders. Ovid’s drug candidate, OV101, is currently in development for the treatment of symptoms of Angelman syndrome and Fragile X syndrome. Ovid is also developing OV935 in collaboration with Takeda Pharmaceutical Company Limited for the treatment of orphan epileptic encephalopathies.
For more information, visit http://www.ovidrx.com/.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements. Forward-looking statements contained in this press release include, without limitation, statements regarding the success of Ovid’s strategic focus or development programs and Dr. French’s contributions to Ovid’s drug development programs, including OV935. Words such as “may,” “believe,” “will,” “expect,” “plan,” “anticipate,” “estimate,” “intend” and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are not guarantees of future performance and involve a number of unknown risks, assumptions, uncertainties and factors that are beyond Ovid’s control. All forward-looking statements are based on Ovid’s expectations and assumptions as of the date of this press release. Actual results may differ materially from these forward-looking statements. Except as required by law, Ovid expressly disclaims any responsibility to update any forward-looking statement contained herein, whether as a result of new information, future events or otherwise.
# # #
Contacts
Investors:
Burns McClellan
Steve Klass, 212-213-0006
Sklass@burnsmc.com
Media:
Pure Communications, Inc.
Katie Engleman, 910-509-3977
katie@purecommunicationsinc.com
New investment will enable Science 37 to scale business operations and enhance technology platform
LOS ANGELES, October 18, 2016 – Science 37 (http://www.science37.com), a technology-enabled clinical research company, today announced that it has completed a $31 million Series B financing. Science 37 is reimagining clinical research by bringing clinical trials to people’s homes, which unlocks access to those beyond a geographical barrier. The investment will help the company grow operations, further development of its novel technology platform that helps accelerate clinical trial research and continue to expand into new therapeutic areas.
The Series B financing was led by Redmile Group, with further funding from Series A lead investors Lux Capital and dRx Capital, as well as a strategic investment from Sanofi Genzyme BioVentures. In addition to financial backing, Sanofi will provide Science 37 with world class technical and strategic guidance through interactions with their international development teams and their affiliates working in the clinical trial ecosystem. As part of the financing, Rob Faulkner, Managing Director at Redmile Group, will join the Science 37 Board of Directors.
Science 37 offers end-to-end decentralized clinical trial services, allowing sponsors and researchers to streamline trial setup, increase the speed of recruitment, improve patient retention and offer exceptional data quality. Science 37 aims to reduce the time and cost of running clinical trials by using technology to improve the patient experience and simplify patient recruitment and retention. Today, the pharmaceutical and medical device industries spend approximately $50 billion annually on clinical trials. Up to 90 percent of these trials face cost overruns and delays. On average, clinical trials can take 6-9 years to complete and represent as much as 40 percent of total therapy development costs. Bottlenecks include identification of investigation sites, trial site start-up, and patient recruitment. Nearly 50 percent of research sites underperform on patient enrollment targets, causing trial delays or even failure.
“The clinical trial process represents significant cost and risk in the development cycle for new therapeutics and medical devices,” said incoming director, Rob Faulkner at Redmile Group. “Science 37 brings a revolutionary approach to the clinical trial process and our investment underscores our confidence in the company’s ability to deliver faster and less expensive clinical trials.”
Introducing a new model to streamline clinical trials
Today’s clinical trial model is built around trial sites, each of which draws from a local geographic area. Up to 70 percent of potential patients live outside of these local areas. Science 37 replaces physical sites with a single “metasite™” and brings the clinical trial directly to the patient’s home. This means that the patient is allowed to retain their existing doctor while benefiting from reduced travel time and access to quality healthcare via telemedicine or doctor home visits when appropriate. Patients with rare diseases or those eligible for precision medicine can gain access to the same trials as those close to traditional sites. This accelerates biomedical discovery and reduces clinical trial costs. The metasite™ is powered by a cloud-based mobile research platform called Network Oriented Research Assistant, (NORA™), that connects investigators and patients, safely and securely.
"Our first investment in Science 37 proved it possible to transform clinical trials and partner with top pharma, biotech and med device companies," said Adam Goulburn, Ph.D., partner at Lux Capital. "Now we’re thrilled to deepen our commitment, putting patients first and using technology to speed life-saving discoveries and cut wasteful costs out of what is today an unquestioned, archaic process."
"Science 37 has demonstrated that its new approach to clinical trial management can drive dramatic improvements for the biomedical industry," said Lucian Iancovici, M.D., partner at dRx Capital. "With our continued investment, Science 37 will have greater capacity to pursue new partnerships and help organizations increase the likelihood of successful clinical trials to find new cures faster."
“We are excited to expand our investment mandate in the digital health space and to start this new initiative with Science 37, a leading company applying transformative digital techniques to clinical trial design and enrollment,” said Bernard Davitian, Managing Director at SGBV. “This investment is fully aligned with Sanofi’s digital strategy and provides an opportunity for us to play an active role in this area of the digital revolution.”
The availability of technology is empowering patients with more information, increasing their role in decision making and reshaping their expectations from healthcare companies.
"We started with the simple question, ‘What is best for patients?’ and built our technology and new clinical trial operating model around the answers. With this latest Series B funding, we will be able to create an even greater impact and achieve our mission of unlocking patient access to clinical trials. Our investors understand how we fit into the larger digital revolution that is forcing healthcare companies to think big, learn fast, adapt successfully, and enable patients to take a more proactive role in their own health," said Noah Craft, M.D., Ph.D., co-founder and CEO of Science 37. "Our early successes last year demonstrated an exponential acceleration of trial speed that has led to incredible demand from biotech and pharmaceutical sponsors. We all share the common goal of bringing new cures to patients faster and this investment will allow us to scale to meet the needs of our clients, while expanding into new therapeutic areas."
About Science 37
Science 37 is a Los Angeles-based technology-enabled clinical research company. The company has created a new clinical trial operating model – the metasite™ – that unlocks access so researchers can find the right patients, and patients can find the right trials. Its Network Oriented Research Assistant, NORA™, is an accelerated patient-centric mobile research platform that connects everyone safely and securely. Science 37 offers end-to-end clinical trial services without geographic limitations, making clinical research faster to accelerate biomedical discovery. Ultimately, Science 37 enables better, faster, people-powered science and brings clinical trials to scale. Learn more at http://science37.com. Follow Science 37 on Twitter @Science_37 or on Facebook @Science37.
About Lux Capital
Lux Capital is a venture firm based in New York City and Silicon Valley investing in counter-conventional, seed and early stage science and technology ventures. The firm manages $700 million in assets across four funds. For more information please see www.luxcapital.com. Follow Lux on Twitter @Lux_Capital.
About dRx Capital
Created in January 2015, dRx Capital is a joint venture investment company launched by Qualcomm (NASDAQ: QCOM) and Novartis (NYSE: NVS). The firm has a US$100M fund dedicated to catalyzing early-stage digital medicine companies. Our team leverages extensive networks in pharma, mobile/IT, and the venture investment community to identify and support promising entrepreneurs. For more information please see www.drxcapital.com. Follow dRx on Twitter @dRxCapital.
About Sanofi-Genzyme BioVentures
Sanofi-Genzyme BioVentures (SGBV) is the corporate venture capital arm of Sanofi. SGBV invests in early stage companies with promising new products that may be future Sanofi pipeline candidates as well as integrated care opportunities. Today, SGBV has assembled a portfolio of direct equity investments in a variety of promising innovative life-science companies. SGBV is an important component of Sanofi’s broader global strategy to invigorate external innovation. For more information, visit: http://sanofigenzymebioventures.com/.
Media Contacts:
Science 37
Ary Hosseinkhani
ary@science37.com
925-519-6553
Partner Sanofi commences dosing in peanut allergy Phase 1 trial leveraging GLAAS
SEATTLE and SOUTH SAN FRANCISCO, Calif., Sept. 28, 2016 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on cancer, today announced the application of its GLAAS discovery platform in Sanofi's Phase 1 clinical trial evaluating a novel therapeutic candidate for the treatment of peanut allergy. The trial follows an exclusive license agreement with Immune Design to discover, develop and commercialize products to treat a peanut allergy utilizing the company's GLAASTM discovery platform.
Sanofi's Phase 1 trial is designed to evaluate the safety of the product candidate in healthy volunteers. Under a previous collaborative research arrangement, Sanofi and IMDZ generated significant preclinical data demonstrating that certain formulations of GLA, the core of the GLAAS platform, when given prophylactically or therapeutically, can shift the immune responses in a way that may result in significant protection and reduction from allergy symptoms. Immune Design has received an undisclosed milestone payment for start of the trial, and is eligible to receive future development and commercialization milestones, as well as tiered royalties on sales of approved products.
"This is the first expansion of the GLAAS platform into allergic diseases, adding to the existing clinical programs in cancer and infectious diseases," said Carlos Paya, M.D., Ph.D., President and Chief Executive Officer of Immune Design. "The GLAAS mechanism of action is designed to help correct imbalances related to immune dysfunction that leads to allergic diseases. The progress of this program into clinical development and GLAAS' robust preclinical data package supports the potential to leverage GLAAS in additional programs in other allergic disease indications."
About Food and Peanut Allergy
The incidence of food allergies is increasing worldwide in both developed and undeveloped countries, and especially in children.1 Globally, experts believe 220-250 million people may suffer from food allergies.1 In the United States alone, as many as 15 million people have food allergies,2 with allergic reactions resulting in an emergency room visit every three minutes and averaging more than 200,000 emergency room visits per year.3 Peanut allergen is the most prevalent, representing approximately 25% amongst food-allergic children in the U.S.4
About GLAAS
Immune Design's GLAAS platform works in vivo and is based on a small synthetic molecule called GLA, or glucopyranosyl lipid A. GLA selectively binds to the TLR4 receptor and causes potent activation of dendritic cells (DCs) leading to the production of cytokines and chemokines that drive a Th1-type immune response. When GLA is accompanied by an antigen and injected into a patient, the combination is taken up by DCs and leads to the production and expansion of immune cells called CD4 T helper lymphocytes with a Th1 phenotype. These CD4 T cells play a key role in boosting pre-existing CTLs that are specific to the same antigen, neutralize a Th2 phenotype, and provide help to other immune cells and natural killer cells that are also important in the overall immune response. GLA can also be used to induce local and systemic immune responses against cancer by directly injecting it into tumors, where it induces a pro-inflammatory state of the tumor microenvironment. Immune Design believes that GLAAS product candidates have the potential to target multiple types of cancer, as well as infectious, allergic and autoimmune diseases. GLAAS-based product candidates have now been evaluated in over 1400 subjects in Phase 1 and Phase 2 trials demonstrating an acceptable safety profile.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the two-pronged focus of Immune Design's ongoing immunooncology clinical programs, are the product of its two synergistic discovery platforms, ZVex® and GLAASTM, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Cautionary Note on Forward-looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the progress, timing and results of clinical trials and the potential receipt of future milestone payments and royalties on sales of approved products. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrolment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, failure of Immune Design's collaborators to support or advance collaborations or product candidates and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
References
- "Food Allergy - A Rising Global Health Problem," World Allergy Week 2013. 8-14 April 2013. http://www.worldallergy.org/UserFiles/file/WorldAllergyWeek2013final.pdf. Accessed online, [August] 2016.
- "Food allergy facts and statistics in the US"; FARE: Food Allergy Research and Education. https://www.foodallergy.org/file/facts-stats.pdf. Accessed on line [September 2016].
- Clark S, Espinola J, Rudders SA, Banerji, A, Camargo CA. Frequency of US emergency department visits for food related acute allergic reactions. J Allergy ClinImmunol. 2011; 127(3):682-683.
- Gupta, Ruchi S. et al. "The Prevalence, Severity, and Distribution of Childhood Food Allergy in the United States," Pediatrics. July 2011; 128(1): e9-e17.
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
NEW YORK--(BUSINESS WIRE)-- Ovid Therapeutics, a privately held biopharmaceutical company based in New York City, announced today that the U.S. Food and Drug Administration has granted orphan drug designation to OV101 for the treatment of patients with Angelman syndrome. OV101 is the first potential therapy to target the disruption of tonic inhibition, a key mechanism that allows the brain to fine-tune neurological signaling and accurately decipher excitatory from inhibitory signals, seen in this disorder.
"Angelman syndrome is a rare, genetic neurological disorder that causes profound developmental and neurologic disabilities. OV101 is the first potential therapy that may address several of the clinical symptoms of Angelman syndrome, such as gait and movement disturbance, anxiety and disrupted sleep patterns," said Dr. Jeremy Levin, chairman and CEO of Ovid Therapeutics. "The granting of orphan drug designation is a significant milestone for the OV101 clinical development program and highlights the high unmet medical need for new therapies that may transform the lives of patients with Angelman syndrome and their families.”
Under the U.S. Orphan Drug Act, the FDA’s Office of Orphan Products Development provides special status and incentives to encourage the development of drugs for diseases affecting fewer than 200,000 people in the U.S. Orphan drug designation conveys up to seven years of marketing exclusivity if the compound receives regulatory approval from the FDA and offers various development incentives, including tax credits related to clinical trial expenses, an exemption from the FDA-user fee and FDA assistance in clinical trial design. The granting of orphan designation does not alter the standard regulatory requirements, timing and process for obtaining marketing approval. Safety and effectiveness of a drug must be established through adequate and well-controlled studies.
About Angelman Syndrome
Angelman syndrome is a rare, genetic disorder that causes developmental disabilities and neurologic problems, such as difficulty speaking, balancing and walking, as well as other symptoms such as anxiety, sleep disturbances and seizures. The most common, genetic cause is a deletion of the ubiquitin protein ligase E3A (UBE3A) gene located on chromosome 15. The first signs of Angelman syndrome are usually developmental delays, such as lack of crawling or walking, seen between the ages of 6-12 months.
Patients with Angelman syndrome lack a key mechanism of the brain known as tonic inhibition, which fine-tunes neurological signals to allow a healthy human brain to clearly conduct messages from one nerve to another and thus maintain a normal balance between excitatory and inhibitory signals without the system being overloaded. Tonic inhibition, a function of the delta (δ)-selective GABA-A receptor, functions to maintain tight control over the level and amount of signaling. When tonic inhibition is “on high,” the brain can decipher signals correctly without being overloaded, but as the level of tonic inhibition is reduced, the brain becomes inundated with signals and loses its deciphering capability.
About OV101
We believe OV101 is the only clinically tested delta (δ)-selective GABA-A receptor agonist and is being developed to help restore tonic inhibition in patients living with Angelman syndrome.
OV101 has the potential to enhance neuronal function to address the pathophysiology of disease. OV101 has been shown in pre-clinical models to normalize tonic inhibition and influence sleep, motor control and cognition, increasing the possibility of improved outcomes for patients.
The STARS trial, a Phase 2, prospective, randomized, double-blind, placebo-controlled clinical trial of OV101 in adults with Angelman syndrome, is currently being initiated in the U.S. in order to assess safety parameters and exploratory efficacy endpoints.
About Ovid Therapeutics Inc.
Ovid Therapeutics Inc. is a privately held, New York-based, biopharmaceutical company committed to transforming the lives of patients with orphan diseases of the brain. Ovid focuses on patients and their unmet medical needs. Using the significant operational, product development, and business development experience of its management team, Ovid aims to become a leading neurology company, with multiple products and a rich pipeline, coupled with compelling research and development. Ovid has raised $80M in financings led by Fidelity Management and Research Company and including Cowen Private Investments, Sanofi-Genzyme BioVentures, Tekla Capital Management, Sphera Global Healthcare Fund, Jennison Associates, Redmile Group, DoubleLine Equity Healthcare Fund, and Cormorant Asset Management, as well as additional blue chip mutual funds and leading life sciences investors.
Contacts
Investors:
Burns McClellan
Steve Klass, 212-213-0006
Sklass@burnsmc.com
or
Media:
Pure Communications, Inc.
Dan Budwick, 973-271-6085
dan@purecommunicationsinc.com
Source: Ovid Therapeutics
Partnership will accelerate discovery of new medicines for neurodegenerative diseases by expanding ability to generate stem cell lines
CAMBRIDGE, Mass., – June 22, 2016 – Yumanity Therapeutics, a company focused on transforming drug discovery for diseases caused by protein misfolding, today announced a discovery collaboration with the New York Stem Cell Foundation (NYSCF) Research Institute, a non-profit organization dedicated to accelerating cures for major diseases through stem cell research. The immediate aim of the partnership is to generate induced pluripotent stem cell (iPSC) lines for use in support of Yumanity Therapeutics’ discovery efforts focused on new medicines for neurodegenerative diseases.
Induced pluripotent stem cells are adult cells that have been genetically reprogrammed and represent an important source of cells that could be used to study the fundamental biology of complex diseases like Alzheimer’s, Parkinson’s and amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease. Since iPSCs are renewable, they are an incredibly useful source of human cells and an increasingly important tool in drug discovery and disease modeling. The New York Stem Cell Foundation’s automated technology, the NYSCF Global Stem Cell Array™, systematically and consistently produces high-quality iPSCs at a scale that enables discovery.
“We are thrilled to be partnering with the NYSCF Research Institute, a world leader in iPSC technology,” said Tony Coles, M.D., chairman and chief executive officer of Yumanity Therapeutics. “The ability to leverage their state-of-the-art capabilities to generate new stem cell lines at scale provides an important step forward in accelerating
Yumanity’s drug discovery efforts.”
“We are excited to partner with Yumanity Therapeutics’ important work in the field of neurodegenerative diseases,” said Susan L. Solomon, co-founder and chief executive officer of NYSCF. “This relationship creates an opportunity to accelerate the kind of game-changing innovation that has always been a priority to us.”
About Yumanity Therapeutics
Yumanity Therapeutics is transforming drug discovery for neurodegenerative diseases caused by protein misfolding. Formed in 2014 by renowned biotech industry leader, Tony Coles, M.D., and protein folding science pioneer, Susan Lindquist, Ph.D., the company is initially focused on discovering disease-modifying therapies for patients with Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis (ALS). Leveraging its three integrated platforms, Yumanity’s innovative new approach to drug discovery and development concentrates on reversing the cellular phenotypes and disease pathologies caused by protein misfolding. For more information, please visit yumanity.com.
About The New York Stem Cell Foundation Research Institute
The New York Stem Cell Foundation (NYSCF) Research Institute is an independent organization accelerating cures and better treatments for patients through stem cell research. The NYSCF Research Institute employs over 45 researchers in New York, and is an acknowledged world leader in stem cell research and in developing pioneering stem cell technologies, including the NYSCF Global Stem Cell ArrayTM. Additionally, NYSCF supports over 85 researchers at other leading institutions worldwide through its Innovator Programs, including the NYSCF – Druckenmiller Fellowships and the NYSCF – Robertson Investigator Awards. NYSCF focuses on translational research in a model designed to overcome the barriers that slow discovery and replaces silos with collaboration. For more information, visit www.nyscf.org
####
Media Contacts:
Pure Communications
Dan Budwick
(973) 271-6085
dan@purecommunicationsinc.com
New York Stem Cell Foundation Research Institute
Maurie Perl
(212) 365-7443
mperl@nyscf.org
LV305, CMB305 and G100 Data at ASCO and June 8 NYC Update Event
SEATTLE and SOUTH SAN FRANCISCO, Calif., June 08, 2016 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on oncology, today announced updated results from clinical trials of three immuno-oncology product candidates demonstrating promising and potentially clinically meaningful anti-tumor immune responses for Immune Design's lead products. This includes data presented at the 52nd Annual Meeting of the American Society of Clinical Oncology (ASCO) annual meeting and involves Immune Design's two distinct immunotherapy applications — the ‘Specific Antigen' and ‘Endogenous Antigen' Approaches.
"The data reflect single agent Phase 1 studies and provide strong rationale for continued development, including our ongoing randomized Phase 2 studies," said Carlos Paya, M.D., Ph.D., President and Chief Executive Officer. "We believe both of our approaches are disruptive and have the potential to impact the immunotherapy treatment landscape."
LV305 & CMB305: Specific Antigen Approach Targeting NY-ESO-1 Positive Tumors — Emerging Profile of Prolonged Survival Benefit
- LV305 Phase 1 single agent trial completed in 24 patients with advanced or metastatic sarcoma cancers expressing NY-ESO-1 (ASCO abstract #3093)
- Median overall survival (OS) has not been reached. One-year survival is 81%.
- Median progression free survival (PFS) is 4.6 months.
- 14 patients (58%) had clinical benefit: One patient (4%) had a late-onset partial response and 13 patients (54%) had stable disease.
- 7/11 patients with pretreatment progressive disease (PD) had SD or PR following LV305.
- Safety profile is very favorable, with only Grade 1/2 adverse effects (AEs).
- l CMB305 Phase 1 single agent trial ongoing in patients with NY-ESO-1 positive soft tissue sarcomas (preliminary analysis of first 14 patients)
- Median OS has not been reached. 93% (13/14 patients) survival to date.
- Median PFS is 5.5 months.
- Best response to date is stable disease (10/14, 71%)
- Safety profile is very favorable, with only Grade 1/2 AE
G100: Intratumoral Immune Activation Approach: Transforming "cold" tumors to "hot" tumors
- G100 single agent and in combination with radiation in patients with Merkel cell carcinoma (ASCO Abstract #3021)
- In final results from 10 patients, G100 produced a 50% overall response rate (ORR) per protocol, including a pathologic complete response (CR) following single agent G100 alone.
- Four patients remain relapse-free in long-term follow up (range 13+ to 27.5+ months).
- Analysis of the tumor microenvironment (TME) in G100-treated patients demonstrates the transformation of a "cold" to a "hot" tumor: increase of innate immune molecules that favor immune cell chemotaxis; increased NK cells and M1 macrophage markers; and dendritic cell antigen presentation. In addition, trafficking of CD4 and CD8 T cells from the stroma into the tumor bed was observed.
- These changes in the TME were most prominent in the G100 responding patients.
- No treatment-related AEs where observed; all AEs were grade 1/2.
Immune Design Company Update June 8
Immune Design will host a Company Update Event: "Progress, Platforms and Paths Forward" on Wednesday, June 8, at 5 p.m. Eastern. Presenters will include: David Baltimore, Ph.D., President Emeritus, California Institute of Technology; Nobel Laureate; Francine M. Foss, M.D., Professor of Medicine, Yale Cancer Center, and Robert G. Maki, M.D., Ph.D., Professor of Medicine, Pediatrics and Orthopaedics, Mt. Sinai Hospital; Director of Translational Research, Sarcoma Alliance for Research Through Collaboration.
A live webcast will be available online from the investor relations page of the company's corporate website at http://ir.immunedesign.com/events.cfm. After the live webcast, an archive of the presentation will be available on the company website for 30 days.
Investors and analysts who would like to attend the Immune Design event in New York should contact Jennifer Cortes at jennifer.cortes@immunedesign.com.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the two-pronged focus of Immune Design's ongoing immunooncology clinical programs, are the product of its two synergistic discovery platforms, ZVexTM and GLAASTM. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
shari.annes@immunedesign.com
650-888-0902
SEATTLE and SOUTH SAN FRANCISCO, Calif. and EMERYVILLE Calif,, May 09, 2016 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on oncology, and Gritstone Oncology, a private cancer immunotherapy company developing next-generation, personalized cancer therapeutics, today announced a clinical collaboration for development of novel, personalized immunotherapies combining both companies' leading technologies.
The collaboration will involve the application of Immune Design's ZVexTM discovery platform with Gritstone's proprietary genomics and proteomics platform for identification of patient-specific tumor antigens to develop neoantigen-based immunotherapies. Immune Design and Gritstone will be jointly responsible for development activities, with an initial likely focus in non-small cell lung cancer. The first clinical trial is expected to commence in 2017.
"The emerging tumor neoantigen field holds great potential for the successful application of cancer immunotherapies, and we are pleased to be working with Gritstone, a company that we believe is a pioneer in the field," said Carlos Paya, M.D., Ph.D., president and chief executive officer of Immune Design. "Having validated our two platforms in clinical trials targeting conserved tumor antigens, we believe their application to patient specific tumor antigens is a natural next step."
For the first trial of their technologies, the companies are evaluating combining the Gritstone and Immune Design neoantigen vaccine with a checkpoint inhibitor, to optimize the vaccine-induced immune response at several levels and maximize the likelihood of clinical efficacy.
"We are excited to work with Immune Design and their novel immunotherapy approach," said Andrew Allen, M.D., Ph.D., co-founder, president and chief executive officer of Gritstone Oncology. "There is good evidence that viral vectors are one of the most effective means of generating high titer CD8+ T cells that recognize encoded antigens, and so this is a logical move for our company, as our neoantigen prediction platform starts to deliver immune targets for individual patients with lung cancer."
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight chronic diseases. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the primary focus of Immune Design's ongoing immuno-oncology clinical programs, are products of its two synergistic discovery platforms, ZVexTM and GLAASTM, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
About Gritstone Oncology
Gritstone Oncology is a privately-held cancer immunotherapy company developing next-generation personalized cancer therapeutics. Gritstone brings together distinguished scientific founders, an experienced and diverse management team, a seasoned and successful board of directors, and deep financial backing to tackle fundamental challenges at the intersection of cancer genomics, immunology, and immunotherapy design. The company's initial goal is to identify and deploy therapeutic neo-antigens from individual patients' tumor to develop novel treatments for lung cancer. Gritstone Oncology is headquartered in the San Francisco Bay Area with certain key functions located in Cambridge, MA. The company launched in October 2015 with a Series A financing of $102 million from leading blue-chip biotechnology investors, including Versant Ventures, The Column Group and Clarus Ventures. More information can be found at www.gritstoneoncology.com.
Immune Design Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend," "likely," "believe," "evaluate" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the timing of initiation, progress, scope and outcome of clinical trials for Immune Design's product candidates, the reporting of clinical data regarding Immune Design's product candidates and the timing of initiation, scope and outcome of the collaborative efforts of Immune Design and Gritstone. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, failure of Immune Design's collaborators to support or advance collaborations or product candidates and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Immune Design Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Immune Design Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
Gritstone Media Contact
Michele Parisi
mparisi@forwardhealthinc.com
925-429-1850
Establishes Potential for the Treatment of Pollen and Peanut Allergies
Data Presented at American Academy of Allergy, Asthma & Immunology (AAAAI) Annual Meeting
SEATTLE and SOUTH SAN FRANCISCO, March 04, 2016 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on oncology, today announced the presentation of data demonstrating the potential of its GLAAS platform to modify allergic immune responses present in pollen and peanut allergies. Results from these studies were highlighted at the 2016 American Academy of Allergy, Asthma & Immunology (AAAI) Annual Meeting in Los Angeles.
Immune Design presented data highlighting the ability of GLA, a synthetic TLR4 agonist and the core of the GLAAS platform, to modify the abnormal allergic immune response observed in peripheral blood from patients with pollen allergies. Specifically, the research demonstrated that GLA significantly decreased the level of Th2 cytokines, IL-5 and IL-13, and increased the level of Th1 cytokines, IFNγ and IL-12, to grass pollen allergen in the peripheral blood mononuclear cells from allergic donors. Th2 cytokines promote the development of an allergic inflammatory response in people with allergies. Th1 cytokines can counterbalance Th2 immune responses and potentially aid in the treatment of allergic diseases.
Abstracts for the 2016 AAAAI Annual Meeting are available online at http://www.jacionline.org/issue/S0091-6749%2815% 29X0003-0 (search "GLA" by article title).
"These presentations add to the large and growing body of data supporting the potential of the GLAAS platform for the treatment of chronic diseases beyond cancer, where Immune Design is focused, including allergy," said Jan ter Meulen, MD, PhD, Chief Scientific Officer at Immune Design. "Investigational agents leveraging GLAAS in oncology are currently in clinical development in randomized Phase 2 studies in soft tissue sarcoma and Non-Hodgkin's lymphoma. Via selective external collaborations and licenses, we are expanding its development in other novel diseases such as peanut food allergy and infectious diseases such as respiratory syncytial virus."
About GLAAS
Immune Design's GLAAS platform works in vivo and is based on a small synthetic molecule called GLA, or glucopyranosyl lipid adjuvant. GLA selectively binds to the TLR4 receptor and causes potent activation of dendritic cells (DCs) leading to the production of cytokines and chemokines that drive a Th1-type immune response. When GLA is accompanied by an antigen and injected into a patient, the combination is taken up by DCs and leads to the production and expansion of immune cells called CD4 T helper lymphocytes with a Th1 phenotype. These CD4 T cells play a key role in boosting preexisting CTLs that are specific to the same antigen, neutralize a Th2 phenotype; and provide help to other immune cells and natural killer cells that are also important in the overall immune response. GLA can also be used to induce local and systemic immune responses against cancer by directly injecting it into tumors, where it induces a pro-inflammatory state of the tumor microenvironment. Immune Design believes that GLAAS product candidates have the potential to target multiple types of cancer, as well as infectious, allergic and autoimmune diseases. GLAAS-based product candidates have now been evaluated in over 1400 subjects in Phase 1 and Phase 2 trials demonstrating an acceptable safety profile.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight chronic diseases. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the primary focus of Immune Design's ongoing immuno-oncology clinical programs, are products of its two synergistic discovery platforms, ZVexTM and GLAASTM, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the timing of initiation, progress, scope and outcome of clinical trials for Immune Design's product candidates and the reporting of clinical data regarding Immune Design's product candidates. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, failure of Immune Design's collaborators to support or advance collaborations or product candidates and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902

CAMBRIDGE, Massachusetts, February 11, 2016 – Proteostasis Therapeutics, Inc. today announced the pricing of its initial public offering of 6,250,000 shares of common stock at a public offering price of $8.00 per share, before underwriting discounts and commissions. All of the common stock is being offered by Proteostasis Therapeutics, Inc. In addition, the company has granted the underwriters a 30-day option to purchase up to an additional 937,500 shares of common stock from the company at the public offering price. The company’s shares are expected to begin trading on the NASDAQ Global Market on February 11, 2016 under the ticker symbol “PTI.” The offering is expected to close on February 17, 2016 subject to customary closing conditions.
Leerink Partners and RBC Capital Markets are acting as joint book-running managers. Baird and H.C. Wainwright & Co. are acting as co-managers.
A registration statement relating to the securities being sold in the offering was declared effective by the Securities and Exchange Commission on February 10, 2016. This offering is being made only by means of a prospectus. Copies of the final prospectus relating to this offering may be obtained by contacting: Leerink Partners LLC, c/o Attention: Syndicate Department, One Federal Street, 37th Floor, Boston, MA 02110, or by email at syndicate@leerink.com, or by phone at (800) 808-7525, ext. 6142; or RBC Capital Markets, LLC, Attention: Equity Syndicate, 200 Vesey Street, 8th Floor, New York, NY 10281, Telephone: (877) 822-4089, Email: equityprospectus@rbccm.com.
This press release shall not constitute an offer to sell or the solicitation of an offer to buy, nor shall there be any sale of these securities in any state or jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction.
Media Contact:
Dan Budwick, Pure Communications
dan@purecommunicationsinc.com
973-271-6085
Investor Contact:
Luke Heagle, Pure Communications
luke@purecommunicationsinc.com
910-726-1372
Investment to Accelerate New Drug Discovery and Target Identification for Alzheimer’s, Parkinson’s and Amyotrophic Lateral Sclerosis
Cambridge, Mass. – February 10, 2016 – Yumanity Therapeutics, a company focused on transforming drug discovery for diseases caused by protein misfolding, today announced a $45 million Series A financing from leading life sciences investors. The financing is being led by Fidelity Management & Research Company with participation by Redmile Group, Alexandria Venture Investments, Biogen, Sanofi-Genzyme BioVentures and Dolby Family Ventures. The Series A financing will accelerate efforts to advance Yumanity Therapeutics’ proprietary platforms aimed at identifying novel therapies to treat neurodegenerative diseases caused by protein misfolding including Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig’s disease. The accumulation of misfolded proteins is believed to play a central role in the initiation and progression of virtually all neurodegenerative diseases.
“We are delighted to have the support of such an accomplished group of investors,” said Tony Coles, M.D., chairman and chief executive officer of Yumanity Therapeutics. “This financing is a testament to the work done over the last year building and validating our platforms, as well as the quality and capability of the company’s scientific team. These proceeds will allow us to devote the resources necessary for our research efforts as we work to discover new drugs for these devastating illnesses. We are strongly positioned to continue executing on the science with the goal of helping the millions of people suffering from neurodegenerative diseases.”
In conjunction with the financing, John Cox, executive vice president of therapeutic operations, Biogen and Joel Marcus, chairman, chief executive officer and founder, Alexandria Real Estate Equities, will join the Yumanity Therapeutics board of directors. Dr. Coles and Jeff Kelly, PhD, chairman, department of molecular and experimental medicine, Scripps Research Institute, will continue to serve on the Yumanity Therapeutics board of directors with Dr. Coles serving as chairman.
“Yumanity Therapeutics brings a skilled, passionate team and unique strategy to targeting the underlying protein pathology of neurodegenerative diseases,” said Bernard Davitian, vice president and managing director of Sanofi-Genzyme BioVentures. “We look forward to working together as investors with Yumanity Therapeutics to advance its discovery platform and make headway against these historically challenging diseases that represent an area of significant medical need.”
It is estimated that more than 55 million people worldwide suffer from neurodegenerative diseases, with no currently approved disease-modifying therapies available.1,2,3 As modern therapeutic interventions increase life expectancy, the number of patients suffering from these diseases is expected to double every 20 years.1,2 Global costs for treating these diseases are currently estimated at $818 billion and expected to grow to more than $1 trillion by 2030.1,2,3
About Yumanity Therapeutics
Yumanity Therapeutics is transforming drug discovery for neurodegenerative diseases caused by protein misfolding. Formed in 2014 by renowned biotech industry leader, Tony Coles, M.D., and protein folding science pioneer, Susan Lindquist, Ph.D., the company is initially focused on discovering disease-modifying therapies for patients with Alzheimer’s disease, Parkinson’s disease and amyotrophic lateral sclerosis (ALS). Leveraging its three integrated platforms, Yumanity’s innovative new approach to drug discovery and development concentrates on reversing the cellular phenotypes and disease pathologies caused by protein misfolding. For more information, please visit yumanity.com.
About Protein Misfolding
DNA is the foundational code for all proteins. The linear information in DNA is first “decoded” into linear strands of amino acids. These strands must then fold in a very precise, highly complex way to form proteins with distinct shapes and functions. When this folding goes awry, critical functions are lost and, even worse, renegade proteins can set off cascades of destruction, causing brain cells to malfunction and die.
Protein misfolding plays a major role in virtually all neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s disease and ALS. While the proteins that misfold in each of these diseases are different, they share this general problem. The cellular stresses caused by protein misfolding lead to aggregation, or clumping of proteins which forms sticky deposits in the brain cells themselves, or in brain tissue, resulting in nerve cell damage and, ultimately, cell death. However, for each of the diseases, as the various “culprit proteins” misfold, they damage nerve cells in very different ways, interacting with different cellular components and causing distinct pathways of cell destruction.
- UBS, Alzheimer’s Report, July 2014; Industry Research.
- Alzheimer’s Disease International, World Alzheimer Report 2015
- Parkinson’s Disease Foundation. http://www.pdf.org/en/parkinson_statistics
CONTACTS:
Yumanity Therapeutics
Dan Budwick
Pure Communications
(973) 271-6085
dan@purecommunicationsinc.com
New Data Support the Continued Development of Both CMB305 and G100
SEATTLE and SOUTH SAN FRANCISCO, Calif., Feb. 09, 2016 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on oncology, today reported positive topline data from three ongoing Phase 1 oncology studies that support continued development of its two primary in vivo immuno-oncology product candidates, CMB305 and G100.
CMB305: First-in-class Prime-boost Immunotherapy Targeting NY-ESO-1 Tumors
Data from a completed first-in-human dose-escalation study and an early subset of patients from an expansion study of CMB305 as a single agent in patients with cancers expressing the NY-ESO-1 tumor antigen revealed:
- CMB305 was safe, without dose-limiting toxicities, as reviewed by an independent data safety monitoring board (DSMB);
- A significant subset of CMB305-treated patients had NY-ESO-1-specific CD8 T cell responses that were generated or increased after therapy;
- Patients who did respond immunologically had a greater degree of antigen-specific T cell response than that previously reported in a Phase 1 study of LV305 alone, which is consistent with the intent of the prime-boost approach; and
- Preliminary clinical benefit in the form of progression-free rate (PFR) was observed in patients with soft tissue sarcoma.
LV305: Novel Vector Delivering NY-ESO-1 RNA Specifically to Dendritic Cells in vivo Maintains Safety and Immunogenicity with Improved Clinical Benefit Profile
Data from the expansion study following the previously-reported dose escalation study of LV305 in patients with tumors expressing NY-ESO-1 revealed:
- A consistently favorable safety profile, as reviewed by an independent DSMB;
- A consistent immune response rate; and
- An improved clinical benefit profile.
G100: Intratumoral Administration of aTLR4 Agonist Significantly Modifies the Tumor Microenvironment (TME) and Maintains Clinical Benefit
New data from the completed pilot trial of G100 with local radiation in patients with Merkel cell carcinoma revealed:
- Safety was consistent with that originally reported, demonstrating an acceptable profile alone or in combination with local radiation;
- G100 significantly altered the TME, causing inflammation and transforming tumors to a "hot" state in G100 responding patients; and
- Clinical benefit remained constant with the full patient set.
Abstracts for each of these three Phase 1 studies have been submitted for presentation at the American Society of Clinical Oncology (ASCO) Annual Meeting (June 3-7, 2016). If afforded the opportunity to present, it is the company's intent to work with the Principal Investigator for each product candidate to present a more complete data set at the Conference.
"The accumulating data, including this new set, clearly supports proceeding with the clinical development of our two first products, CMB305 and G100, each activating the anti-tumor immune response by targeting a predefined tumor antigen or neo-antigens, respectively," said Carlos Paya, M.D., Ph.D, President and Chief Executive Officer of Immune Design. "The initiation of randomized studies in which we are combining our two products with inhibitors of the PD-1/L1 axis through our collaborations with Genentech and Merck will provide the evidence as to how novel products that activate the immune system and aim to make tumors "hot" add or synergize with check-point inhibitors."
Additional Information on Immune Design's Distinct Immuno-oncology Approaches
About CMB305
CMB305 is an immuno-oncology product candidate that involves the sequential dosing of two active agents, LV305 and G305. LV305 is a hybrid vector from the ZVex™ discovery platform that specifically targets dendritic cells (DCs) in vivo and delivers the RNA for NY-ESO-1, enabling the DCs to express the entire tumor antigen and potentially induce a diverse set of CTLs targeting NY-ESO-1 in tumors. G305, in contrast, is designed to boost the CTL response via the induction of antigenspecific CD4 "helper" T cells. G305 consists of recombinant NY-ESO-1 protein formulated with a proprietary synthetic small molecule called glucopyranosyl lipid A (GLA), the novel TLR4 agonist at the core of the GLAAS™ platform. CMB305 is intended to be an "off-the shelf" therapy that does not require patient-specific manufacturing or ex vivo manipulation of patient samples. Immune Design has conducted prior studies to establish the safety and individual immunologic activity of LV305 and G305. CMB305 is currently being evaluated in a Phase 1B trial in patients with locally advanced, relapsed or metastatic solid cancers whose tumors express NY-ESO-1 and a randomized Phase 2 trial of CMB305 combined with Genentech's investigational cancer immunotherapy, atezolizumab (anti-PD-L1), in patients with soft tissue sarcoma, pursuant to a collaboration with Genentech.
About G100
G100 is Immune Design's intratumoral Immune Activation approach to treating cancer and is expected to directly activate dendritic and other antigen presenting cells near the tumor, which may enhance the function of pre-existing cytotoxic T lymphocytes (CTLs) and create both a local and systemic immune response against neo-antigens. G100 is a product of the company's GLAAS platform and recently completed a Phase 1 study in patients with Merkel cell carcinoma. G100 also has potential therapeutic utility in any accessible tumor and will be investigated in combination with local radiation and Merck's anti-PD-1 agent, KEYTRUDA®, in a randomized study in patients with follicular Non-Hodgkin lymphoma, pursuant to a collaboration with Merck.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the primary focus of Immune Design's ongoing immunooncology clinical programs, are products of its two synergistic discovery platforms, ZVex and GLAAS, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the timing of initiation, progress, scope and outcome of clinical trials for Immune Design's product candidates and the reporting of clinical data regarding Immune Design's product candidates. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, failure of Immune Design's collaborators to support or advance collaborations or product candidates and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
Shari.Annes@immunedesign.com
650-888-0902
Company Plans to Double in Size in 2016 as It Aggressively Expands Its Scientific Workforce
CAMBRIDGE, MA, January 26, 2016 – Unum Therapeutics, a company developing a universal cellular immunotherapy to treat multiple cancers, today announced its move into new office and laboratory facilities in Cambridge, MA. The company’s new headquarters, located at 200 Cambridge Park Drive, was designed to help facilitate Unum’s anticipated growth to more than 80 employees in the near future. As part of the program to recognize Unum’s recent progress and advancing drug development portfolio, Robert Coughlin, President & CEO of the Massachusetts Biotechnology Council, Mike Kennealy, Assistant Secretary for Business Growth at the Executive Office of Housing & Economic Development, and Angus McQuilken, Vice President for Marketing & Communications for the Massachusetts Life Sciences Center participated in a ribbon cutting ceremony earlier today.
“I congratulate Unum Therapeutics on the opening of their new facility, and on the work they are doing to find new treatments and advancements in healthcare,” said Governor Baker. “I look forward to their continued contributions to job creation and innovation here
in the Commonwealth.”
“We believe that our ACTR technology platform fuels one of the industry’s most promising immunotherapy drug development programs,” said Chuck Wilson, PhD, President & CEO of Unum Therapeutics. “To reach our full potential, we need to continue recruiting top-level talent. This move allows us to expand our hiring efforts within the world-class Cambridge, Massachusetts life sciences hub, to house a larger team of scientists, and to continue to leverage our proximity to leading research and academic institutions and healthcare companies. Unum is excited to be joining a growing biotech community in West Cambridge.”
Unum designed its new 33,000 square feet of space specifically to foster continued collaboration, including state-of-the-art laboratories and an open office design. Employees can easily access the centrally located facility via public transportation at The Alewife Reservation (including access to the MBTA Red Line) and local highways, including Route 2 and Route 16.
Coming just a year after the company’s launch, this move follows several major milestones for Unum including initiation of a Phase 1 study in B-cell malignancies in Singapore, a $65M Series B financing, and a major strategic collaboration with Seattle Genetics. Unum evolved from the LabCentral incubator program in Kendall Square, where it was located prior to the Cambridge Drive facility.
“We are pleased to celebrate the grand opening of Unum’s new headquarters in Cambridge,” said Travis McCready, President & CEO of the Massachusetts Life Sciences Center. “This new facility will allow the company to continue to grow and make important contributions to cancer therapeutics, one of the core strengths of our Commonwealth's life sciences ecosystem. The company’s growth is just one early return on the MLSC’s investment in the construction of LabCentral.”
“We are thrilled to celebrate Unum’s growth as a great example of the opportunity presented to companies in the Massachusetts ecosystem,” said Robert K. Coughlin, President & CEO of MassBio. “As a company with great science and great talent that has taken advantage of the efficiencies provided by an incubator and the network provided by active participation at MassBio, Unum has provided a clear path to success for other startups here.”
About Unum Therapeutics
Unum Therapeutics uses proprietary T-cell engineering technology in combination with tumor-targeting antibodies to activate the body’s own immune system to fight cancer. Unum’s lead program, based on its Antibody-Coupled T-cell Receptor (ACTR) technology, recently entered Phase 1 clinical testing to assess safety and efficacy. FierceBiotech recently named Unum Therapeutics as one of 2015’s Fierce15 biotechnology companies, designating it as one of the most promising private biotechnology companies in the industry. The company is headquartered in Cambridge, MA. For more information, visit http://www.unumrx.com.
Media Contact:
Mike Beyer
Sam Brown Inc.
+1-312-961-2502
mikebeyer@sambrown.com
Designation for LV305 and G305
SEATTLE and SOUTH SAN FRANCISCO, Calif., Jan. 08, 2016 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on oncology, today announced that the U.S. Food and Drug Administration (FDA) has granted Orphan Drug Designation for LV305 and G305 for the treatment of soft tissue sarcoma. LV305 and G305 are the complementary agents that comprise CMB305, Immune Design's "prime boost" cancer immunotherapy product candidate.
Orphan Drug Designation is granted by the FDA Office of Orphan Drug Products to products that treat rare diseases. The FDA defines rare diseases as those affecting fewer than 200,000 people in the United States. Orphan Drug Designation provides the sponsor certain benefits and incentives, including a period of marketing exclusivity for the first marketing application, if regulatory approval is received for the designated indication, potential tax credits for certain activities and waiver of certain administrative fees.
CMB305 is a cancer immunotherapy product candidate that involves the sequential dosing of LV305 and G305. LV305 is a hybrid vector from the ZVex™ discovery platform that specifically targets dendritic cells (DCs) in vivo and delivers the RNA for NY-ESO-1, enabling the DCs to express the entire tumor antigen and potentially induce a diverse set of CTLs targeting NY-ESO-1 in tumors. G305, in contrast, is designed to boost the CTL response via the induction of antigen-specific CD4 "helper" T cells. G305 consists of recombinant NY-ESO-1 protein formulated with a proprietary synthetic small molecule called glucopyranosyl lipid A (GLA), the novel TLR4 agonist at the core of the GLAAS™ platform. CMB305 is intended to be an "off-the shelf" therapy that does not require patient-specific manufacturing or ex vivo manipulation of patient samples. Immune Design has conducted prior studies to establish the safety and individual immunologic activity of LV305 and G305. CMB305 is currently being evaluated in a Phase 1B trial in patients with locally advanced, relapsed or metastatic solid cancers whose tumors express NY-ESO-1 and a randomized Phase 2 trial of CMB305 combined with Genentech's investigational cancer immunotherapy, atezolizumab (MPDL3280A; anti-PD-L1) in patients with soft tissue sarcoma, pursuant to a collaboration with Genentech.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the primary focus of Immune Design's ongoing immunooncology clinical programs, are products of its two synergistic discovery platforms, ZVex and GLAAS, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the timing of initiation, progress, scope and outcome of clinical trials for Immune Design's product candidates and the reporting of clinical data regarding Immune Design's product candidates. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, failure of Immune Design's collaborators to support or advance collaborations or product candidates and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
Proceeds Will Accelerate Advancement of Lead mTORC1 Modulator Program Into Clinical Evaluation
CAMBRIDGE, Mass., December 18, 2015 – Navitor Pharmaceuticals, Inc., a biopharmaceutical company developing novel medicines targeting the activation of mTORC1, today announced the completion of a $33 million Series B financing. Proceeds from the financing will be used to accelerate the development of Navitor’s pipeline of first-in-class selective modulators of the mTORC1 pathway that plays a key role in a range of chronic diseases of aging as well as rare disorders. The Series B funding will enable Navitor to advance a lead program into clinical trials with the objective of demonstrating initial efficacy and safety. The company will also continue to develop additional pipeline candidates and augment its proprietary drug discovery platform.
The Series B round was led by new investor Brace Pharma Capital, LLC, a strategic investment company formed by EMS S.A., the largest pharmaceutical company in Brazil, and high net worth biotech investors, and includes participation by new investors Remeditex Ventures, Sanofi-Genzyme BioVentures, and an undisclosed individual investor. Existing investors Polaris Partners, Atlas Venture, Johnson & Johnson Innovation – JJDC, Inc. and SR One, Ltd. also participated in the financing.
“We are excited to welcome three exceptional new investors to our syndicate and have the continued support of our existing key investor group. We believe this financing reflects excitement for our innovative therapeutic approach of selectively targeting mTORC1 and recognizes our significant progress over the past year in advancing our lead small molecule programs,” said George P. Vlasuk, PhD, President and Chief Executive Officer of Navitor. “As we move forward with discovering first-in-class medicines, we are delivering on our mission to create next generation therapies that significantly improve the lives of patients affected by chronic diseases of aging and other disorders associated with dysregulation of mTORC1 activation. The additional capital from this financing gives Navitor the necessary resources to advance our drug discovery efforts toward the demonstration of clinical efficacy.”
The Series B financing will enable the advancement of Navitor’s pipeline of novel drug compounds that target the mTORC1 activation pathway towards the demonstration of clinical efficacy and safety, opening up the potential for expanded therapeutic opportunities characterized by mTORC1 dysregulation that are currently underserved by available treatments. The pipeline includes novel therapeutics that have the potential to either “turn up” or “turn down” nutrient and growth factor-responsive cellular pathways to restore normal mTORC1 function. By selectively targeting and controlling aberrant cellular signaling mediated by the mTORC1 pathway, Navitor’s therapeutics are designed to rebalance these signals and restore normal function and impact the underlying biology of multiple diseases that are the result of mTORC1 dysregulation, including metabolic, neurodegenerative, immune/autoimmune and musculoskeletal diseases, as well as several rare disorders.
In conjunction with the financing, Vinzenz Ploerer, President & CEO of Brace Pharma Capital, will join the Navitor Board of Directors. Mr. Ploerer stated “Navitor has assembled an outstanding team and is executing a sound strategy that has yielded promising results against multiple unprecedented targets leading to the dysregulation of mTORC1 activity.”
Raymond Schinazi, PhD, DSc, the Founder of Pharmasset and a member of both the Board of Directors and the Scientific Advisory Board of Brace Pharma Capital, commented further “Selective modulation of mTORC1 activation has the potential to generate first-in-class innovative therapeutics for multiple indications with significant unmet need. Navitor has assembled a world-class team that has the experience and vision to create tremendous value for patients.”
Barry Burgdorf, Chief Operating Officer of Remeditex Ventures, who will also join the Navitor Board of Directors, added “Remeditex invests in biotech companies that are hard at work on advancing breakthrough science with the promise to alleviate major health burdens and which are led by a management team of vision and creativity. Navitor matches this profile completely.”
About mTORC1
The mTOR (mechanistic target of rapamycin) kinase exists in two multi‐protein complexes within the cell, called mTORC1 and mTORC2. Both complexes are critical signaling nodes that regulate multiple cellular functions including metabolism, growth and response to changes in the cell’s environment. mTORC1 responds to and integrates the cell’s response to nutrient availability and growth factors and plays a key role in protein synthesis and cellular growth. As a critical regulatory pathway, mTORC1 is often dysregulated in multiple diseases across several important therapeutic areas. While several approved drugs (rapamycin and related allosteric mTORC1 inhibitors) target the broad mTOR pathway for certain specific disease applications, the use of these first generation drugs has been limited since they inhibit both mTORC1 and mTORC2, leading to undesirable side effects when used chronically. Navitor’s therapeutics are designed to selectively modulate the cellular signals that are aberrant in disease processes caused by the dysregulation of mTORC1 activity without inhibiting mTORC2.
About Navitor
Navitor Pharmaceuticals, Inc., is a biopharmaceutical company developing novel medicines by targeting cellular nutrient signaling pathways. The company’s proprietary drug discovery platform targets mTORC1, which responds to and integrates the cell’s response to nutrient availability and plays a key role in protein synthesis and cellular growth. Navitor’s therapeutics are designed to selectively modulate the cellular signals that are aberrant in disease processes caused by the dysregulation of mTORC1 activation to address a wide range of diseases, including metabolic, neurodegenerative, autoimmune and musculoskeletal diseases, as well as age-related immune suppression and several rare disorders. The company’s founding intellectual property is based on the groundbreaking discoveries related to the mTORC1 pathway and nutrient signaling mechanisms by Dr. David Sabatini at The Whitehead Institute for Biomedical Research. The company is backed by leading financial and corporate investors, including Polaris Partners, Atlas Venture, Johnson & Johnson Innovation – JJDC, Inc., SR One, Ltd., Brace Pharma Capital, Remeditex Ventures and Sanofi-Genzyme Bioventures. For more information, please visit www.navitorpharma.com.
CONTACT:
Kathryn Morris
The Yates Network
845-635-9828
kathryn@theyatesnetwork.com
Promotes Ben Munoz, Ph.D. to Senior Vice President, Drug Discovery and Po-Shun Lee, M.D. to Executive Vice President and Chief Medical Officer
CAMBRIDGE, Massachusetts, December 11, 2015 – Proteostasis Therapeutics, Inc. (PTI), a biopharmaceutical company developing small molecule therapeutics to treat diseases caused by defects in protein processing, announced today that Ben Munoz, Ph.D. has been promoted to Senior Vice President, Drug Discovery. PTI also announced the promotion of Po-Shun Lee, M.D. to Executive Vice President. Dr. Lee will also continue to serve as the company’s Chief Medical Officer. Markus Haeberlein will transition out of his role as Chief Scientific Officer to pursue other career opportunities.
“We are delighted to promote Ben and Po-Shun, who have demonstrated their leadership, competence and diligence in the evolution of the company. I am confident each will continue to contribute meaningfully to the company’s success as we advance our cystic fibrosis development program and expand our pipeline,” said Meenu Chhabra, President and Chief Executive Officer of PTI.
In his new role, Dr. Munoz will direct all of the company's drug discovery research activities and operations. He will also be responsible for expanding the pipeline with new drug candidates by overseeing activities related to hit identification, lead optimization, drug candidate selection as well as all related aspects of chemistry, manufacturing and controls.
In his expanded role, Dr. Lee will lead all research and development activities related to cystic fibrosis and will serve as a key member of the executive leadership team.
Ben Munoz, Ph.D.
Dr. Munoz joined PTI in 2013. Since joining PTI, he has been responsible for managing all internal, external and outsourcing chemistry efforts from hit identification through development candidate selection.
In his former role at the Broad Institute, Dr. Munoz was responsible for directing and coordinating the oncology and infectious disease platform, and Molecular Library Probe Production Center Network (MLPCN) effort. He worked to design and implement a phenotypic screening platform which led to potent molecules with therapeutic potential in oncology, neuroscience, infectious and neglected diseases. He also worked at Merck Research Laboratories in Boston and San Diego.
Po-Shun Lee, M.D.
Dr. Lee joined PTI in 2014. He is a pulmonary and critical care physician with extensive experience in biopharmaceutical research and development in both the industry and academia. He has led the cystic fibrosis team and strategic selection process of the Company’s proprietary CFTR amplifier compounds as the Company proceeds toward clinical development.
Prior to joining PTI, Dr. Lee led the cystic fibrosis and asthma programs from early development to proof of concept at the Novartis Institute for Biomedical Research. Before joining Novartis, Dr. Lee worked at Vertex Pharmaceuticals where he supported the clinical development and registration of Kalydeco and led a CFTR corrector program to positive proof of concept.
About Proteostasis Therapeutics
Proteostasis Therapeutics, Inc. (PTI) is developing disease-modifying therapeutics for diseases of protein processing. By combining the DRT™ platform, a phenotypic screening approach based on the use of functionally pertinent cellular assays and disease relevant models, PTI identifies highly selective drug candidates that modulate the proteostasis imbalance in the cell. In addition to its multiple wholly-owned programs in cystic fibrosis, PTI has formed collaborations with Biogen New Ventures Inc. to research and identify therapeutic candidates for neurodegenerative disease and with Astellas Pharma Inc. to research and identify therapies targeting the Unfolded Protein Response (UPR) pathway. http://www.proteostasis.com/
Contact:
Janet Smart
Proteostasis Therapeutics, Inc.
617-225-0096 Ext. 2128
Janet.Smart@proteostasis.com
Media | Proteostasis Therapeutics
CAMBRIDGE, Massachusetts, November 20, 2015 – Proteostasis Therapeutics, Inc. (PTI), acompany developing small molecule therapeutics to treat diseases caused by defects in protein processing, announced today the expansion of its cystic fibrosis (CF) drug candidate pipeline to include the addition of a novel triple combination therapy of PTI’s own CFTR amplifiers, correctors and potentiators. PTI’s testing has shown that a triple combination comprising a proprietary corrector and potentiator, currently in late lead optimization stage, and one of PTI’s CFTR amplifiers, can restore the activity of mutant F508del CFTR protein to 80% of normal activity in Ussing chamber assays.
The components of the triple combination were developed internally using the Company’s proprietary Disease-Relevant Translation, or DRT™, platform. The screening assay was optimized to identify novel CFTR modulators that show functional synergy with the Company’s lead drug candidate, PTI-428, which belongs to a novel class of CFTR modulators the Company refers to as “amplifiers”, while restoring chloride currents above levels achieved by existing commercially available products. In Ussing chamber assays, one PTI potentiator, PTI-P271, has shown comparable efficacy with Vertex Pharmaceuticals’ (Vertex) potentiator, ivacaftor, and did not cause F508del CFTR protein destabilization under chronic administration conditions when combined with a PTI corrector. Also in Ussing chamber assays, one PTI corrector, PTI-C1811, has been shown to restore at least 140% of CFTR functional levels relative to Vertex’s corrector lumacaftor, which is currently marketed together with ivacaftor as Orkambi™.
Based on the data generated in the human bronchial epithelial (hBE) cells homozygous for the F508del mutation, the Company believes that the combined use of the three molecules has the potential to restore mutant CFTR function in CF patients homozygous for the F508del mutation to approximately 80% of normal activity. Further, PTI-C1811 and PTI-P271 combined have demonstrated higher levels of CFTR function in vitro than Orkambi™.
“The unique screening set-up allowed us to identify novel chemical moieties with corrector and potentiator properties that act synergistically with our CFTR amplifiers across several CFTR mutation classes” said Meenu Chhabra, President and Chief Executive Officer of PTI. “We are very pleased with the rapid advancement of all of our CF programs, and are confident that we will continue to build on our promising amplifier program to expand the range of treatment options for most CF patients.”
PTI is advancing its CFTR amplifier PTI-428 as its lead clinical development candidate for the treatment of CF and expects to file an IND with the FDA by the end of 2015. The PTI correctors and potentiators are expected to enter clinical development by the middle of 2017.
About Cystic Fibrosis
Cystic fibrosis is a genetic disorder affecting approximately 70,000 to 100,000 people worldwide.
Improvement in disease management protocols and approval of new drugs to treat the symptoms have extended the life expectancy for CF patients. However, CF remains an incurable disease that leads to death.
About Proteostasis Therapeutics
Proteostasis Therapeutics, Inc. (PTI) is developing disease-modifying therapeutics for diseases of protein processing. By combining the DRT™ platform, a phenotypic screening approach based on the use of functionally pertinent cellular assays, with state of the art medicinal chemistry tools, PTI generates highly selective drug candidates that modulate the proteostasis imbalance in the cell. In addition to its multiple wholly-owned programs in CF, PTI has formed collaborations with Biogen Inc. to research and identify therapeutic candidates for neurodegenerative disease and with Astellas Pharma Inc. to research and identify therapies targeting the Unfolded Protein Response (UPR) pathway.
Investor Contact:
Janet Smart
Proteostasis Therapeutics, Inc.
617-225-0096 Ext. 2128
Janet.Smart@proteostasis.com
Study to Evaluate Combination of Immune Design's CMB305 and Genentech's Atezolizumab
SEATTLE and SOUTH SAN FRANCISCO, Calif., Nov. 11, 2015 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on oncology, today announced the start of a randomized Phase 2 trial of CMB305, the company's "prime boost" cancer immunotherapy product candidate, combined with Genentech's investigational cancer immunotherapy, atezolizumab (MPDL3280A; anti-PD-L1) in patients with soft tissue sarcoma.
The open label trial is designed to evaluate the safety and efficacy of CMB305 in combination with atezolizumab versus atezolizumab alone in up to 80 patients with locally advanced, relapsed, or metastatic synovial sarcoma or myxoid/round-cell liposarcoma expressing the NY-ESO-1 cancer testis antigen. The trial is being conducted pursuant to a clinical collaboration with Genentech, a member of the Roche Group, which will provide atezolizumab for the trial.
CMB305 is a "prime-boost" cancer immunotherapy product designed to synergistically induce and expand in vivo cytotoxic T lymphocytes (CTLs) targeting NY-ESO-1 which is found in a broad set of tumors. Specifically, synovial sarcoma and myxoid/round-cell liposarcoma tend to express NY-ESO-1 broadly, which should make them good indications for clinical studies of this antigen-specific immune therapy. Atezolizumab is designed to target PD-L1 expressed on tumor cells and tumorinfiltrating immune cells, preventing it from binding to PD-1 and B7.1 on the surface of T cells. By inhibiting PD-L1, atezolizumab may enable the activation of T cells..
"This trial provides our first opportunity to validate the fundamental hypothesis that combining cutting-edge technologies designed to trigger in vivo anti-tumor CTLs with antagonists of the PD-1/PD-L1 axis should be additive, if not synergistic, and thus could enhance the potential therapeutic benefit to patients," said Carlos Paya, M.D., Ph.D., President and Chief Executive Officer of Immune Design. "We are excited to work with leading sarcoma investigators to advance CMB305 into its first randomized Phase 2 trial and to explore the potential of these two approaches."
About CMB305
CMB305 is a cancer immunotherapy product candidate combining two potentially synergistic agents, LV305 and G305. LV305 is a hybrid vector from the ZVex™ discovery platform that specifically targets dendritic cells (DCs) in vivo and delivers the RNA for NY-ESO-1, enabling the DCs to express the entire tumor antigen and potentially induce a diverse set of CTLs targeting NYESO- 1 in tumors. G305, in contrast, is designed to boost the CTL response via the induction of antigen-specific CD4 "helper" T cells. G305 consists of recombinant NY-ESO-1 protein formulated with a proprietary synthetic small molecule called glucopyranosyl lipid A (GLA), the novel TLR4 agonist at the core of the GLAAS™ platform. CMB305 is intended to be an "offthe shelf" therapy that does not require patient-specific manufacturing or ex vivo manipulation of patient samples. Having established the safety and individual immunologic activity of LV305 and G305 in prior studies, Immune Design initiated a new Phase 1B study of the product candidate CMB305 earlier this year.
About Soft Tissue Sarcoma
Soft Tissue Sarcomas (STS) are malignancies that arise from the soft tissues of the body, such as tissues that connect, support and surround other body structures including muscle, fat, blood vessels, nerves, tendons and the lining of joints.1 In the United States, nearly 12,000 people will be diagnosed and approximately 4,870 are expected to die of STS in 2015.2 There are approximately 50 different types of STS including Liposarcomas and Synovial Sarcomas, which are subtypes affecting fat tissue and tissue around the joints, respectively.3 Myxoid/round cell is a type of liposarcoma that accounts for approximately 30% of all liposarcoma cases.4 Myxoid and round cell liposarcoma and synovial sarcomas have been shown to have high expression of NY-ESO-1, approximately 90% and approximately 60%, respectively.5
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the primary focus of Immune Design's ongoing immuno-oncology clinical programs, are products of its two synergistic discovery platforms, ZVexand GLAAS, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the timing of initiation, progress, scope and outcome of clinical trials for Immune Design's product candidates and the reporting of clinical data regarding Immune Design's product candidates. Many factorsmay cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, failure of Immune Design's collaborators to support or advance collaborations or product candidates and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forwardlooking statements contained herein to reflect any change in expectations, even as new information becomes available.
1 Mayo Clinic. Disease and Conditions: Soft tissue sarcoma.
Available at: http://www.mayoclinic.org/diseases-conditions/soft-tissue-sarcoma/basics/definition/con-20033386. Accessed:November 2015.
2 National Cancer Institute. SEER Stat Fact Sheets: Soft Tissue including Heart Cancer.
Available at: http://seer.cancer.gov/statfacts/html/soft.html. Accessed: November 2015.
3 American Cancer Society. What is a soft tissue sarcoma?
Available at: http://www.cancer.org/cancer/sarcoma-adultsofttissuecancer/detailedguide/sarcoma-adult-soft-tissue-cancer-softtissue-
sarcoma. Accessed: November 2015
4 Orpha.net. Myxoid/round cell liposarcoma.
Available at: http://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=99967. Accessed: November 2015.
5 Endo, M., et al. (April 2015). NY-ESO-1 (CTAG1B) expression in mesenchymal tumors [Abstract]. Modern Pathology, 28, 587- 595.
Available at: http://www.ncbi.nlm.nih.gov/pubmed/25412843. Accessed: November 2015.
CONTACT:
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
Source: Immune Design
News Provided by Acquire Media
Demonstrates Potential of "Prime-Pull" Immunotherapy Approach
SEATTLE and SOUTH SAN FRANCISCO, Calif., Nov. 3, 2015 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on oncology, today announced new preclinical data showing that a dendriticcell targeting lentiviral vector from its ZVex™ immunotherapy platform administered with G100, which contains a potent synthetic TLR4 agonist, synergize with immune check point inhibitors and demonstrate potent local and systemic anti-tumor activity in cancer models. These data are being presented at the 30th Annual Meeting of the Society for Immunotherapy of Cancer (SITC) Conference, National Harbor, Maryland, November 4-8, 2015.
"These findings further illustrate the potential of ZVex-based product candidates and G100 to play an important role in the emerging field of cancer immunotherapy, especially in potential combination therapies with immune checkpoint inhibitors," said Jan ter Meulen, MD, PhD, Chief Scientific Officer at Immune Design. "Mechanistically, it is generally believed that the generation of tumor-specific CD8 T-cells can improve the clinical efficacy of checkpoint inhibitors, especially in patients with insufficient T-cell responses. These preclinical data provide a strong rationale for our ongoing and planned clinical trials which combine LV305, CMB305 and G100, agents from our ZVex and GLAAS discovery platforms, with KEYTRUDA and Atezolizumab."
In the presentations, Immune Design scientists present data showing that in the murine B16 melanoma model, intratumoral injection of G100 improves the trafficking of ZVex-induced antigen-specific CD8 T-cells into tumors and induces systemic antitumor immunity mediated by antigen spreading. This discovery that Immune Design's two platforms appear to "pull" T cells (G100) that were "primed" (ZVex agent) into the tumor potentially opens up new possibilities of enhancing the immunotherapy of solid tumors by changing the tumor microenvironment. In addition, combining G100 or a ZVex agent with both anti-PD1 and anti-PDL1 antibodies demonstrated increased efficacy in this experiment. The presentations are entitled "Intratumoral Injections of G100 (synthetic TLR4 agonist) Increases Trafficking of Lentiviral Vector-induced Antigen-specific CD8 T Cells to the Tumor Microenvironment" and "Checkpoint Inhibitors Synergize with Therapeutic Platforms, ZVex™ and GLAAS™ by Enhancing Lentiviral Vector-induced Tumor-specific Immunity and Adjuvant-mediated Anti-tumor Efficacy."
These abstracts will be published in the Journal for ImmunoTherapy for Cancer on November 4, 2015, and the posters will be posted on the publications page of the Immune Design website following presentation at the conference.
About G100
G100 is a product candidate generated from the company's GLAASTM discovery platform, and includes a specific formulation of Glucopyranosyl Lipid A (GLA), a synthetic, toll-like receptor-4 (TLR-4) agonist. G100 is part of Immune Design's intratumoral immune activation, or 'Endogenous Antigen' approach to treating cancer, which leverages the activation of dendritic cells, T cells and other immune cells in the tumor microenvironment to potentially create a robust immune response against the tumor's preexisting diverse set of antigens. Preclinical and clinical data have demonstrated the ability of G100 to activate dendritic cells in tumors and to increase antigen-dependent systemic humoral and cellular Th1 immune responses. A Phase 1 study of G100 in patients with Merkel cell carcinoma (MCC) recently completed enrollment, and Immune Design presented data at the 2015 American Society of Clinical Oncology (ASCO) Annual Meeting. In the first eight patients in MCC study, G100 had an acceptable safety profile and the combined therapy of G100 followed by radiation and/or surgery resulted in an objective response rate (ORR) of 50%. A second Phase 1 trial is planned to examine intratumoral administration of G100 with intravenous administration of KEYTRUDA® (pembrolizumab), Merck's anti-PD-1 therapy, in patients with follicular non-Hodgkin's lymphoma receiving local radiation. In addition to an evaluation of the safety of the combination, the study will assess the response in both injected and non-injected lesions.
About ZVex™
ZVex is Immune Design's discovery platform designed to activate and expand the immune system's natural ability to create tumor-specific cytotoxic T cells (CTLs) in vivo.
The ZVex delivery system uses a re-engineered virus to carry genetic information of a tumor antigen selectively to dendritic cells (DCs) in the skin. This ultimately results in the creation of CTLs designed to kill tumor cells bearing that same specific tumor antigen.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the primary focus of Immune Design's ongoing immuno-oncology clinical programs, are products of its two synergistic discovery platforms, ZVexTM and GLAASTM, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Forward Looking Statement:
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the timing of initiation, progress, scope and outcome of clinical trials for Immune Design's product candidates and the reporting of clinical data regarding Immune Design's product candidates. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, failure of Immune Design's collaborators to support or advance collaborations or product candidates and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forwardlooking statements contained herein to reflect any change in expectations, even as new information becomes available.
CONTACT:
Contact for Immune Design:
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206.769.9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
SEATTLE and SOUTH SAN FRANCISCO, Calif., Oct. 29, 2015 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on oncology, today highlighted the application of its GLAASTM discovery platform in MedImmune's Phase 2 clinical trial of MEDI7510. MEDI7510 is an investigational agent for the prevention of respiratory syncytial virus (RSV) under development by MedImmune, the global biologics research and development arm of AstraZeneca. MEDI7510 is composed of MedImmune's RSV sF antigen plus GLA, a synthetic molecule licensed from Immune Design's GLAAS discovery platform.
This stems from an existing agreement in which Immune Design granted MedImmune an exclusive license to use the GLAAS platform to develop and commercialize vaccines in two different infectious disease indications, one of which is RSV.
The Phase 2, double-blind, randomized, placebo-controlled study (NCT02508194) is designed to assess the efficacy of MEDI7510 for the prevention of acute RSV-associated respiratory illness in older adults. The study will also evaluate the safety and immunogenicity of MEDI7510 or placebo and immune response to MEDI7510 in Season 1 and Season 2. The trial is expected to enroll approximately 1,900 adult subjects, 60 years or older, globally.
"It's rewarding to see MEDI7510 continue to advance through clinical development," said Carlos Paya, M.D., Ph.D., President and Chief Executive Officer at Immune Design. "The field of RSV vaccines has been challenging. A small molecule that activates TLR4 and drives a Th1-type of immune response should be ideally suited to overcome the Th2-prone activity of RSV antigens, which can result in severe lung pathology. We are hopeful of the potential benefit MEDI7510 may bring to older adults."
ABOUT RSV
Respiratory Syncytial Virus (RSV) is a common virus that can cause upper and lower respiratory infections, including colds, bronchitis and pneumonia. RSV is increasingly recognized as an important cause of respiratory infections in adults, particularly affecting the elderly, immunocompromised, and those with underlying chronic cardiopulmonary disease. For example, RSV is estimated to infect 5%-10% of nursing home residents per year, with rates of pneumonia and death of 10%-20% and 2%-5%, respectively (Falsy, Clin Microbiol Rev 2000). Currently no vaccine is available for RSV and induction of a Th1 type immune response is viewed as an important feature for any successful vaccine candidate.
About GLAAS
Immune Design's GLAAS platform works in vivo and is based on a small synthetic molecule called GLA, which stands for glucopyranosyl lipid A. GLA selectively binds to the TLR4 receptor and causes potent activation of dendritic cells (DCs). When GLA is accompanied by an antigen and injected into a patient, the combination is taken up by DCs and leads to the production and expansion of immune cells called CD4 T helper lymphocytes. These CD4 cells play a key role in boosting the anti-tumor immune response by expanding the number and function of existing CTLs that are specific to the same antigen and providing help to other immune cells, including B lymphocytes that are the precursor to antibodies and natural killer cells that are also important in the overall immune response. In addition to infectious diseases, Immune Design believes that GLAAS product candidates have the potential to target multiple types of cancer and allergic diseases.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the primary focus of Immune Design's ongoing immuno-oncology clinical programs, are products of its two synergistic discovery platforms, ZVexTM and GLAASTM, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Forward Looking Statement:
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the timing of initiation, progress, scope and outcome of clinical trials for Immune Design's product candidates and the reporting of clinical data regarding Immune Design's product candidates. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, failure of Immune Design's collaborators to support or advance collaborations or product candidates and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forwardlooking statements contained herein to reflect any change in expectations, even as new information becomes available.
CONTACT:
Contact for Immune Design:
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206.769.9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
-Former SVP at Millennium/Takeda Fills Newly Created Position to Advance Unum’s Expanding Immunotherapy Clinical Program
CAMBRIDGE, MA, October 21, 2015 – Unum Therapeutics, a company developing a universal cellular immunotherapy to treat multiple cancers, announced today that it has named Michael J. Vasconcelles, M.D., to serve as the company’s Chief Medical Officer, effective immediately. He is responsible for defining the strategic vision and leading execution of the company’s clinical efforts to develop its universal Antibody-Coupled T-cell Receptor (ACTR) product. In this capacity, Dr. Vasconcelles oversees a growing portfolio of combination therapies that incorporate marketed, partnered, and proprietary antibodies.
“We are embarking on a new and exciting phase of our company’s expansion, progressing rapidly into clinical development and expanding a pipeline of programs,” said Chuck Wilson, Ph.D., President & CEO of Unum Therapeutics. “It’s a true win for us to attract an industry veteran with the breadth of experience and track record of accomplishment that Mike brings to our company. He has the vision to understand the potential of our ACTR technology, the skills to work successfully with our development partners, and the means to quickly and safely deliver the technology to the benefit of patients.”
Dr. Vasconcelles most recently served as the Senior Vice President, Global Head, Oncology Therapy Area Unit, Takeda Pharmaceuticals International Co. where he was accountable for the oncology research and development strategy and progression of the oncology portfolio from candidate selection through life cycle management. During Dr. Vasconcelles’ tenure, Takeda’s oncology portfolio averaged approximately 12 investigational agents or marketed products, including new Investigational New Drug Applications (INDs) or IND-equivalents and several assets each in early clinical development pre-proof of concept, pivotal development, and/or commercialized with ongoing clinical investigation intended for expanded indications.
“Immunotherapy continues to make great strides in the treatment of cancer patients with limited options, and I am very excited by Unum’s vision of a single cellular immunotherapy that may be used to treat many types of cancers,” said Dr. Vasconcelles. “It is an optimal time for me to join the company as it embarks on the critical phase of clinical translation. I am looking forward to leading the team that plays an integral part in the clinical value proposition for the platform.”
Prior to his leadership post at Takeda, Dr. Vasconcelles has held several positions at Genzyme Corporation and Sanofi, including Global Therapeutic Area Head, Oncology and Transplantation, and Head, Personalized Medicine and Companion Diagnostics, respectively.
Dr. Vasconcelles currently holds positions as a Clinical Instructor in Medicine at Harvard Medical School and as a staff physician at two prestigious institutions: Dana-Farber Cancer Institute and Brigham & Women’s Hospital in Boston, Massachusetts.
He received his B.A. from Northwestern University, and his M.D. from Northwestern University’s Feinberg School of Medicine. Dr. Vasconcelles is a member of numerous professional societies, including the American Society of Clinical Oncology and the American Society of Hematology. He is a member of the board of the Personalized Medicine Coalition, and a grant reviewer for the United States National Institutes of Health (NIH). He has been the recipient of several major research grants from the NIH and has been published in peer-reviewed journals such as the Journal of Clinical Oncology, Journal of Biological Chemistry, Molecular and Cellular Biology and the Journal of Immunotherapy.
About Unum Therapeutics
Unum Therapeutics uses proprietary T-cell engineering technology in combination with tumor-targeting antibodies to activate the body’s own immune system to fight cancer. Unum’s lead program, based on its Antibody-Coupled T-cell Receptor (ACTR) technology, recently entered Phase 1 clinical testing to assess safety and efficacy. The company is headquartered in Cambridge, MA. For more information, visit www.unumrx.com.
NEW YORK--(BUSINESS WIRE)-- Ovid Therapeutics Inc. (Ovid), a privately held biopharmaceutical company focused on developing therapies for rare and orphan diseases of the brain, announced today the appointment to its Scientific Advisory Board (SAB) of Jerome B. Zeldis, M.D., Ph.D., Chief Executive Officer of Celgene Global Health and Chief Medical Officer of Celgene Corporation.
Dr. Zeldis is noted for his exceptional and successful experience in drug development. Prior to his current role at Celgene, Dr. Zeldis has served as Celgene’s Senior Vice President of Clinical Research and Medical Affairs. Prior to joining Celgene in 1997, Dr. Zeldis was Associate Director of Clinical Research at Sandoz Research Institute where he helped develop Zelnorm® (tegaserod) and Director of Medical Development at Janssen Pharmaceutical Research Institute where he worked on Prepulsid® (cisapride). He currently serves as Chairman of the board of Semorex Corporation and has additional board positions at Alliqua, Bionor Pharma, PTC Therapeutics, Soligenix, and Trek Therapeutics. He was Assistant Professor of Medicine at Harvard Medical School, Associate Professor of Medicine at University of California, Davis, Clinical Associate Professor of Medicine at Cornell Medical School, and Professor of Clinical Medicine at the Robert Wood Johnson Medical School. Dr. Zeldis received BA and MS degrees from Brown University, and M Phil, MD, and PhD degrees from Yale University.
“We are delighted and privileged to welcome Jerry to our Scientific Advisory Board,” said Dr. Jeremy Levin, Chief Executive Officer and Chairman of Ovid. “Jerry’s advice and insight will be extremely valuable as we enter the clinic.”
Dr. Zeldis commented, “Ovid is pursuing an ambitious approach of tackling rare neurological diseases with significant unmet needs. With this goal, the company has assembled a world-class leadership and is employing a science-based approach to target new pathways that are involved in these diseases. I look forward to working closely with Ovid’s team.”
About Ovid Therapeutics Inc.
Ovid Therapeutics Inc. is a privately held, New York-based, biopharmaceutical company committed to transforming the lives of patients with rare and orphan diseases of the brain. Ovid focuses on patients and their unmet medical needs. Using the significant operational, product development, and business development experience of its management team, Ovid aims to become a leading neurology company, with multiple products and a rich pipeline, coupled with compelling research and development. Ovid recently completed a substantially oversubscribed $75 Million Series B financing led by Fidelity Management and Research Company and including Cowen Private Investments, Sanofi-Genzyme BioVentures, Tekla Capital Management, Sphera Global Healthcare Fund, Jennison Associates (on behalf of certain clients), Redmile Group, and Cormorant Asset Management, as well as additional blue chip mutual funds and leading life sciences investors.
For more information on Ovid, please visit http://www.ovidrx.com.
View source version on businesswire.com: http://www.businesswire.com/news/home/20151008006425/en/
Contacts
Burns McClellan, on behalf of Ovid Therapeutics
Justin Jackson, 212-213-0006, ext. 327
jjackson@burnsmc.com
Source: Ovid Therapeutics Inc.
CAMBRIDGE, MA--(Marketwired - Oct 8, 2015) - Proteostasis Therapeutics, Inc. (PTI), a company developing novel therapeutics to treat diseases caused by defects in protein processing, today announced the presentation of data on the Company's CFTR amplifier program and its potential for use in combination therapies for cystic fibrosis (CF) at the 29th Annual North American Cystic Fibrosis Conference in Phoenix, Arizona.
In the Company's oral presentation titled "CFTR Amplifiers Are a New Class of CFTR Modulators", PTI reported an observed increase in cystic fibrosis transmembrane conductance regulator (CFTR) modulating activity in human bronchial epithelial (hBE) cells, when its compounds were used as stand-alone treatment, and evidence that these compounds can further enhance CFTR mediated current when used in combination with existing clinical-stage correctors.
Because mutations in the CFTR gene cause CF, CFTR amplifiers can represent a new class of CFTR modulating agents with potential therapeutic use in the treatment of this severe and incurable condition. CFTR amplifiers enhance the effect of existing CFTR modulators, such as potentiators and correctors. The CFTR amplifiers have demonstrated potential to be effective across CFTR mutation classes in preclinical studies and such results form the basis for PTI's strategy to develop a broad-acting combination therapy able to serve CF patients.
"We are very pleased with the clinical potential of the CFTR amplifier compounds that double the activity of the most effective combination of clinical-stage correctors in the gold-standard HBE cell assay, not only for the most common mutation, F508del/F508del, but in other mutations found in the cystic fibrosis population as well," said Meenu Chhabra, President and Chief Executive Officer of PTI. "We are confident that we will continue to build on our promising preclinical results to advance our products toward clinical trials."
The Company is developing PTI-428 as a new CFTR amplifier for the treatment of CF. PTI-428 has demonstrated pharmacologic properties amenable to oral dosing. A twenty-eight day, non-GLP preclinical toxicology study testing multiple dose groups of PTI-428 in non-human primates demonstrated a favorable safety and tolerability profile for clinical development. PTI will advance PTI-428 as its lead clinical development candidate for the treatment of CF and expects to file an IND with the FDA by the end of 2015.
Additionally, PTI has initiated a new preclinical program in chronic obstructive pulmonary disease (COPD) with another one of its CFTR amplifiers, PTI-130. COPD is characterized by shortness of breath, coughing and increased mucus formation, which can be a significant contributor to morbidity. PTI-130 may represent a novel treatment approach due to its ability to increase CFTR-mediated ion transport in non-CF hBE cells. Targeting CFTR function may potentially improve hydration and restore mucus formation to normal physiological levels in the airway.
PTI's CFTR amplifiers are wholly-owned by PTI and were internally discovered through the company's proprietary Disease-Relevant Translation, or DRT™ platform. PTI-428 and PTI-130 are results of medicinal chemistry optimization of internally discovered active compounds. In tests using HBE cell Ussing functional assays, both drug candidates increased CFTR function and nearly doubled the efficacy of CFTR modulating agents such as correctors and potentiators. These data provide a basis for the clinical exploration of the use of PTI-428 as an add-on therapy to the emerging standard of care (corrector/potentiator combination) to deliver greater benefit to CF patients, and the use of PTI-130 to enhance non-mutant CFTR function in COPD.
About Cystic Fibrosis
Cystic fibrosis is a genetic disorder affecting approximately 70,000 to 100,000 people worldwide. Improvement in disease management protocols and approval of new drugs to treat the symptoms have extended the life expectancy for CF patients. However, CF remains an incurable disease that leads to death.
About Chronic Obstructive Pulmonary Disease
Chronic obstructive pulmonary disease, or COPD, is a type of chronic progressive lung disease characterized by poor airflow. COPD is a leading cause of death worldwide. The main symptoms include shortness of breath, cough and sputum production. There is no known cure for COPD. The current standard of care is therapeutic intervention to alleviate symptoms and delay progression of the disease.
About Proteostasis Therapeutics
Proteostasis Therapeutics, Inc. (PTI) is developing disease-modifying therapeutics for diseases of protein processing. By combining the DRT™ platform, a phenotypic screening approach based on the use of functionally pertinent cellular assays, with state of the art medicinal chemistry tools, PTI generates highly selective drug candidates that modulate the proteostasis imbalance in the cell. In addition to its multiple wholly-owned programs in CF and COPD, PTI has formed collaborations with Biogen Inc. to research and identify therapeutic candidates for neurodegenerative disease and with Astellas Pharma Inc. to research and identify therapies targeting the Unfolded Protein Response (UPR) pathway.
Contact Information
Investor Contact:
Janet Smart
Proteostasis Therapeutics, Inc.
617-225-0096 Ext. 2128
CAMBRIDGE, MA, September 30, 2015 – Unum Therapeutics today announced that it has been named by FierceBiotech as one of 2015’s Fierce 15 biotechnology companies, designating it as one of the most promising private biotechnology companies in the industry.
Unum is a Cambridge, MA-based immunooncology company creating the next generation of engineered cellular therapies. The company has developed an antibody-coupled T-cell receptor (ACTR) that, when combined with tumor-specific antibodies, directs a patient’s T-cells to kill tumor cells. In contrast to other approaches, Unum’s cell therapy is not restricted to a specific antigen and may be useful for attacking a range of different types of cancers.
“We are honored to be recognized by FierceBiotech and to be included in such a strong, promising group of Fierce companies,” said Chuck Wilson, PhD, President & CEO of Unum Therapeutics. “In the past year, we have made significant progress in building our company and bringing our ACTR therapy to patients. We look forward to continuing to advance and expand our pipeline and to realizing the potential of our ACTR therapy in treating patients with hematological and solid tumors.”
The Fierce 15 celebrates the spirit of being “fierce” – championing innovation and creativity, even in the face of intense competition. This is FierceBiotech’sthirteenth annual Fierce 15 selection. A complete list of “Fierce 15” companies can be found online at www.fiercebiotech.com/fierce15.
An internationally recognized daily report reaching a network of over 275,000 biotech and pharma industry professionals, FierceBiotech provides subscribers with an authoritative analysis of the day's top stories. Every year FierceBiotech evaluates hundreds of private companies from around the world for its annual Fierce 15 list, which is based on a variety of factors such as the strength of its technology, partnerships, venture backers and a competitive market position.
About FierceBiotech
FierceBiotech is the biotech industry's daily monitor, an email newsletter and web resource providing the latest biotech news, articles, and resources related to clinical trials, drug discovery, FDA approval, FDA regulation, patent news, pharma news, biotech company news and more. More than 150,000 top biotech professionals rely on FierceBiotech for an insider briefing on the day's top stories. Signup is free at www.fiercebiotech.com/signup.
About Unum Therapeutics
Unum Therapeutics uses proprietary T-cell engineering technology in combination with tumor-targeting antibodies to activate the body’s own immune system to fight cancer. Unum’s lead program, based on its Antibody-Coupled T-cell Receptor (ACTR) technology, recently entered Phase 1 clinical testing to assess safety and efficacy. The company is headquartered in Cambridge, MA. For more information, visit http://www.unumrx.com
September 30, 2015 04:15 PM Eastern Daylight Time
NEW YORK--(BUSINESS WIRE)--Ovid Therapeutics Inc. (Ovid), a privately held biopharmaceutical company focused on developing therapies for rare and orphan diseases of the brain, announced today the appointment of Karen Bernstein, Ph.D., to the Company’s Board of Directors. Dr. Bernstein is widely known and recognized for co-founding and shaping BioCentury Publications Inc. into one of the most highly regarded sources of industrial intelligence and news of the Global BioPharmaceutical Industry.
“Her intellect, perspectives, and deep knowledge of the life science industry will be tremendous additions to our Board’s deliberations. We are privileged that she has joined Ovid’s board.”
"Karen has been instrumental in charting and interpreting the growth and evolution of the biotechnology industry. Her work and that of BioCentury is iconic,” said Dr. Jeremy Levin, Chief Executive Officer and Chairman of Ovid. “Her intellect, perspectives, and deep knowledge of the life science industry will be tremendous additions to our Board’s deliberations. We are privileged that she has joined Ovid’s board.”
Dr. Bernstein co-founded BioCentury in 1992 alongside President and CEO David Flores, with the goal of delivering the essential clinical, regulatory, and finance news shaping the biotechnology and pharmaceutical industries. Under their leadership, BioCentury became a key independent source of intelligence for a global audience of biopharmaceutical executives, investors, regulators, and public policy influencers.
Dr. Bernstein commented, “I have had the great pleasure to know the members of Ovid’s team over many years, and it is very exciting to see this group come together in forming a new company that I believe will make a major difference for patients with rare and orphan diseases of the brain. Recent scientific insights in CNS diseases open new avenues for the development of important new drugs.
Ovid is uniquely suited to become a major player in this area. I very much look forward to being a part of the company’s progress and success.”
Dr. Bernstein stepped down from her role as Editor-in-Chief in August, but will continue serving as BioCentury’s Chairman. In addition to joining Ovid’s board of directors, Dr. Bernstein has also joined the board of Vitae Pharmaceuticals. She currently serves as a trustee of the Keck Graduate Institute of the Applied Life Sciences and is a member of the Keck Graduate Institute School of Pharmacy’s board of advisors. Dr. Bernstein earned a Ph.D. in political science from Stanford University and a B.A. in politics and history from Brandeis University.
About Ovid Therapeutics Inc.
Ovid Therapeutics Inc. is a privately held, New York-based, biopharmaceutical company committed to transforming the lives of patients with rare and orphan diseases of the brain. Ovid focuses on patients and their unmet medical needs. Using the significant operational, product development and business development experience of its management team, Ovid aims to become a leading neurology company, with multiple products and a rich pipeline, coupled with compelling research and development. Ovid recently completed a substantially oversubscribed $75 Million Series B financing led by Fidelity Management and Research Company and including Cowen Private Investments, Sanofi-Genzyme BioVentures, Tekla Capital Management, Sphera Global Healthcare Fund, Jennison Associates (on behalf of certain clients), Redmile Group, and Cormorant Asset Management, as well as additional blue chip mutual funds and leading life sciences investors.
For more information on Ovid, please visit http://www.ovidrx.com.
Contacts
Burns McClellan, on behalf of Ovid Therapeutics
Justin Jackson, 212-213-0006, ext. 327
jjackson@burnsmc.com
Funding Supports Advancement of Therapies for the Treatment of Cystic Fibrosis
CAMBRIDGE, MA--(Marketwired - Sep 8, 2015) - Proteostasis Therapeutics, Inc. (PTI), a company developing novel therapeutics to treat diseases caused by defects in protein processing, today announced that it has secured $37 million in a Series B mezzanine equity financing. The company plans to use the proceeds of this financing to advance its lead product candidate in cystic fibrosis into human clinical studies and continue to expand its product portfolio. The financing was led by Cormorant Asset Management and also included Rock Springs Capital Management. Existing investors New Enterprise Associates, Elan Science One, HealthCare Ventures, Fidelity Biosciences, Novartis Bioventures, Novartis Venture Fund, and Sanofi-Genzyme BioVentures also participated in the financing. Leerink Partners LLC acted as the exclusive placement agent to the company for the offering.
"Cormorant Global Healthcare Fund is pleased to participate in the mezzanine equity financing of PTI. We believe PTI's pipeline of drug candidates has the potential to provide significant benefits to a broad group of cystic fibrosis patients. We are excited to support this experienced management team as they advance a series of compounds through clinical trials. We are happy to join such an innovation-focused investor group and to help PTI achieve its mission to improve the lives of patients with cystic fibrosis," stated Bihua Chen, Managing Member and Chief Executive Officer of Cormorant Asset Management.
"We are very pleased that we were able to attract such a strong investor group to our company at this important time in our development and we welcome the new investors to our existing group of top-tier investors," said Meenu Chhabra, PTI's President and Chief Executive Officer. "As we advance our programs into clinical development, PTI is in a strong position to improve upon present treatments for patients with cystic fibrosis. We are confident that we will continue to build on our promising preclinical results to advance our lead product in to the clinic."
About CFTR Amplifiers
Proteostasis Therapeutics, Inc.'s lead asset represents a new class of agents, called cystic fibrosis transmembrane conductance regulator (CFTR) amplifiers, for the treatment of cystic fibrosis (CF). CFTR amplifiers represent a new drug class able to enhance the effect of known CFTR modulators, such as potentiators and correctors. In preclinical studies this amplifier was effective across CFTR mutation classes and forms the basis for PTI's strategy to seek to develop a broad-acting combination therapy able to serve CF patients with most mutations. The lead asset is fully-owned by PTI and was internally discovered through the company's proprietary Disease-Relevant Translation (DRT™) platform. CF is a genetic disorder affecting approximately 70,000-100,000 people worldwide. Improvement in disease management protocols and approval of new drugs to treat the symptoms have extended the life expectancy for CF patients, which is now approaching 40 years of age. However, CF remains an incurable disease that leads to death.
About Proteostasis Therapeutics
Proteostasis Therapeutics, Inc. is developing disease-modifying therapeutics for diseases of protein processing. By combining the DRT™ platform, a phenotypic screening approach based on the use of functionally pertinent cellular assays, with state of the art medicinal chemistry tools, PTI generates highly selective drug candidates that modulate the proteostasis imbalance in the cell. In addition to its multiple wholly-owned programs in CF, PTI has formed collaborations with Biogen Inc. to research and identify therapeutic candidates for neurodegenerative disease and with Astellas Pharma Inc. to research and identify therapies targeting the Unfolded Protein Response (UPR) pathway.
CONTACT INFORMATION
Investor Relations Contact:
Chris Brinzey
Westwicke Partners
339-970-2843
Chris.Brinzey@westwicke.com

- Proceeds from financing to enable advancement of multiple product pipeline of antigen-specific immunotherapies and vaccines
- Phase 2 study of lead immunotherapeutic candidate, SEL-212, in gout planned to start in 2016
WATERTOWN, Mass. – September 8, 2015 – Selecta Biosciences, Inc., a clinical stage biotechnology company developing a novel class of targeted antigen-specific immune therapies, announced today that it has completed a $38 million Series E equity financing. Proceeds from the financing will be used to advance multiple product candidates from Selecta’s Synthetic Vaccine Particle (SVP) platform, which creates antigen-specific immunotherapeutics, with priority implementation of the clinical program for Selecta’s lead immunotherapeutic candidate, SEL-212, which is designed to be the first non-immunogenic biologic therapy for gout. The largest investor in this Series E round is OrbiMed, a leading institutional healthcare investor, and includes new investors, Sanofi-Genzyme BioVentures, Ridgeback Capital Management, Osage University Partners, AJU IB Investment and Sphera Global Health Care Fund, along with participation from all of Selecta’s existing investors.
The Series E financing will allow Selecta to complete the on-going Phase 1 clinical program for SEL-212 in refractory and tophaceous gout, and then initiate a multi-dose ascending phase 2 study which is planned to start in 2016. Proceeds from the financing will also be used to advance the company’s preclinical pipeline of antigen-specific immunotherapeutics toward clinical development, which includes candidates that are designed to prevent the life threatening rejections that are a key unmet medical need of gene therapies and Factor VIII therapies. Selecta is additionally developing first-in-class therapies for an undisclosed food allergy, celiac disease and type 1 diabetes, all in collaboration with Sanofi, whose venture capital division Sanofi-Genzyme BioVentures is an investor in this financing round.
“Selecta has demonstrated leadership in the field of immune therapeutics and has made strong progress with a completely novel class of products addressing multiple therapeutic areas with significant unmet needs,” said Carl Gordon, PhD, CFA, Partner at OrbiMed. “We have great confidence in the high caliber management team and the rigorous science and clinical program behind SEL-212 and other immunotherapeutic candidates in Selecta’s robust pipeline. We look forward to supporting Selecta’s plans to open up new therapeutic opportunities and to bring innovative medicines to patients in need across a number of important disease areas, such as inhibition of immunogenicity of biologic therapies, treatment of allergies and treatment of autoimmune diseases.”
“With this oversubscribed round that includes a balance of crossover, strategic and venture investors, we are very pleased to see this strong support from the investor community for our antigen-specific immunotherapies. We now have the resources and investor base that will see us through the Phase 2 clinical development of SEL-212, the first non-immunogenic biologic therapy for gout, and the extension of our clinical pipeline into first ever non-immunogenic gene therapies and therapeutic biologics,” noted Werner Cautreels, PhD, President and CEO at Selecta.
In addition to the crossover and strategic investors who participated in this round, Selecta is backed by top-tier venture investors, including Polaris Venture Partners, Flagship Ventures, NanoDimension, Rusnano and Leukon Investments, who all participated in this round.
About Selecta
Selecta Biosciences, Inc. is a clinical-stage biotechnology company developing novel drugs that use immune modulating nanomedicines to generate targeted antigen-specific immune responses to prevent and treat disease. Selecta’s proprietary Synthetic Vaccine Particle (SVP) platform creates a novel paradigm in immunotherapeutics and vaccines, enabling completely new applications while offering the potential of improved efficacy and safety profiles.
Selecta’s immunomodulatory SVPs can induce antigen-specific immune tolerance, enabling them to be applied in a variety of therapeutic areas with large unmet medical need. The company is focused on three key near-term applications: inhibition of immunogenicity of biologic therapies, treatment of allergies and treatment of autoimmune diseases. Immunogenicity adversely affects the safety and efficacy profile for many biological therapies, and is known to have caused the termination of a number of promising biological therapies in clinical development. Selecta’s SVP is a product engine that has the potential to unlock the full therapeutic value of biologic therapies.
Through proprietary products and collaborations with leading pharmaceutical companies and research organizations, Selecta is building a pipeline of product candidates to address unmet medical needs in serious and chronic diseases. Selecta Biosciences, Inc. is based in Watertown, Massachusetts, USA. For more information, please visit www.selectabio.com.
# # #
For Selecta media:
Kathryn Morris
The Yates Network
+1-845-635-9828
kathryn@theyatesnetwork.com
For Selecta investors:
Stephanie Ascher
Stern Investor Relations, Inc.
+1-212-362-1200
stephanie@sternir.com
NEW YORK — August 11, 2015 — Ovid Therapeutics Inc. (Ovid), a privately held biopharmaceutical company focused on developing therapies for rare and orphan diseases of the brain, announced today that it has completed a Series B financing totaling $75 million. Fidelity Management and Research Company led the investment syndicate, which included Cowen Private Investments, Sanofi-Genzyme BioVentures, Tekla Capital Management, Sphera Global Healthcare Fund, Jennison Associates (on behalf of certain clients), Redmile Group, and Cormorant Asset Management, as well as additional blue chip mutual funds and leading life sciences investors. Existing Ovid investors, including DoubleLine Equity Healthcare Fund, LLC, also participated in the financing.
“Over the last year, we have rapidly executed on our strategy with the support of our investors, patients, their families, foundations, physicians, and corporate partners,” said Dr. Jeremy Levin, Chief Executive Officer and Chairman of Ovid. “Looking to the future, this financing is an important next step toward our goal to build Ovid into a leading neurology company.”
Ovid plans to use the proceeds from the Series B financing to advance its product candidates, including OV101 for Angelman Syndrome and Fragile X Syndrome. Phase 2 clinical testing is expected to commence in both patient groups in 2016. In addition, Ovid plans to use the funds to advance several other internal compounds into clinical testing, and to further expand its pipeline.
“We are pleased to secure this syndicate of top-tier investors,” said Dr. Yaron Werber, Chief Financial Officer. “This oversubscribed financing will provide significant latitude for business and corporate development initiatives and allow us to further accelerate Ovid’s programs.”
Cowen and Company served as the sole placement agent for the financing, and Hogan Lovells US LLP served as counsel to Ovid.
About Ovid Therapeutics Inc.
Ovid Therapeutics Inc. is a privately held, New York-based, biopharmaceutical company committed to transforming the lives of patients with rare and orphan diseases of the brain. Ovid focuses on patients and their unmet medical needs. Using the significant operational, product development and business development experience of its management team, Ovid aims to become a leading neurology company, with multiple products and a rich pipeline, coupled with compelling research and development.
For more information on Ovid, please visit http://www.ovidrx.com.
Contacts
Burns McClellan, on behalf of Ovid Therapeutics
Justin Jackson,
212-213-0006, ext. 327
jjackson@burnsmc.com
Phase 1 Trials to Evaluate Investigational Agents G100 or LV305 Combined With Merck's KEYTRUDA(R) (pembrolizumab)
SEATTLE, SOUTH SAN FRANCISCO, Calif. and KENILWORTH, N.J., Aug. 10, 2015 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ) today announced it has entered into clinical collaboration agreements through subsidiaries of Merck (NYSE:MRK), known as MSD outside of the United States and Canada, to evaluate the safety and efficacy of two Immune Design immuno-oncology investigative agents, G100 and LV305, separately combined with KEYTRUDA® (pembrolizumab), Merck's anti-PD-1 therapy, in Phase 1 trials in patients with non-Hodgkin's lymphoma (NHL) and melanoma, respectively.
The first clinical trial will examine intratumoral administration of G100 with intravenous administration of KEYTRUDA in patients with follicular NHL receiving local radiation. In addition to an evaluation of the safety of the combination, the study will assess the response in both injected and non-injected lesions. The second clinical trial in melanoma will evaluate safety and response to the combination of LV305 and KEYTRUDA in patients who have not yet responded to treatment with KEYTRUDA alone after three months of treatment.
Immune Design's G100 and LV305 investigational agents are designed to work in vivo and activate the immune system via the induction and/or expansion of anti-tumor CD8 T cells. They are intended to be "off-the-shelf" therapies, in contrast to other Tcell approaches that require individualized ex vivo manipulation. G100 is a potent toll-like receptor-4 (TLR4) agonist designed to generate a robust anti-tumor immune response when administered directly to the tumor micro-environment. LV305, in contrast, is designed to activate the immune system through the in vivo generation of cytotoxic T cells (CTLs), initially against a specific tumor-associated antigen, NY-ESO-1. Immune Design is studying LV305 primarily as part of CMB305, a prime boost approach currently in a Phase 1 expansion trial.
"There is great potential to expand the potential of immunotherapy through combination approaches that will stimulate and enhance the immune system in order to mount the strongest response against cancer," said Carlos Paya, M.D., Ph.D, President and Chief Executive Officer of Immune Design. "Immune Design has two distinct approaches in oncology, and we look forward to collaborating with Merck to evaluate the potential of combining each of G100 and LV305 with KEYTRUDA in these areas of medical need."
"Our understanding of the immune system's role and its impact in the treatment of cancer continues to grow," said Dr. Roger M. Perlmutter, president, Merck Research Laboratories. "This collaboration with Immune Design adds to a broad clinical program designed to explore the role of KEYTRUDA in innovative immuno-oncology combinations - and underscores our commitment to advance the care of patients with cancer."
About G100
G100 is a product candidate generated from the company's GLAASTM discovery platform, and includes a specific formulation of Glucopyranosyl Lipid A (GLA), a synthetic, toll-like Receptor-4 (TLR-4) agonist. G100 is part of Immune Design's intratumoral immune activation, or 'Endogenous Antigen' approach to treating cancer, which leverages the activation of dendritic cells, T cells and other immune cells in the tumor microenvironment to potentially create a robust immune response against the tumor's preexisting diverse set of antigens. Preclinical and clinical data have demonstrated the ability of G100 to activate dendritic cells in tumors and to increase antigen-dependent systemic humoral and cellular Th1 immune responses. In addition to the study planned under this collaboration, a Phase 1 study of G100 in patients with Merkel cell carcinoma (MCC) recently completed enrollment, and Immune Design presented data at the 2015 American Society of Clinical Oncology (ASCO) Annual Meeting, the poster for which can be accessed on the company's website. In the first eight patients in MCC study, G100 has an acceptable safety profile and a fifty percent (50%) objective response rate per protocol.
About LV305
LV305, generated from Immune Design's ZVexTM platform, is designed to activate the immune system through the in vivo generation of cytotoxic T cells (CTLs) initially against a specific tumor-associated antigen, NY-ESO-1. LV305 is part of Immune Design's 'Specific Antigen' approach, which drives the in vivo generation of a strong, antigen-specific CTL response against selected antigens present in a tumor. Preclinical tests have demonstrated the ability of LV305 to reduce tumor growth of NYESO- 1-expressing tumors, increase production of antigen-specific CD8 cells, and significantly improve the survival of tumor bearing animals. LV305 is the first step in Immune Design's novel prime-boost approach to immuno-oncology, which includes combination with G305, generated from the GLAAS platform, to expand CTLs and potentially generate a potent, durable immune response. Immune Design announced positive data from a Phase 1 study of LV305 at the 2015 ASCO Annual Meeting, the poster for which can be accessed on the company's website. In that study, LV305 caused either a de novo or statisticallysignificant increase in antigen-specific CD8 T cells in 80% of the six evaluable mid- and high-dose patients. Immune Design is primarily studying LV305 as part of CMB305, a prime boost approach.
About KEYTRUDA® (pembrolizumab)
KEYTRUDA (pembrolizumab) is a humanized monoclonal antibody that blocks the interaction between PD-1 and its ligands, PD-L1 and PD-L2. By binding to the PD-1 receptor and blocking the interaction with the receptor ligands, KEYTRUDA releases the PD-1 pathway-mediated inhibition of the immune response, including the anti-tumor immune response.
KEYTRUDA is indicated in the United States at a dose of 2 mg/kg administered as an intravenous infusion over 30 minutes every three weeks for the treatment of patients with unresectable or metastatic melanoma and disease progression following ipilimumab and, if BRAF V600 mutation positive, a BRAF inhibitor. This indication is approved under accelerated approval based on tumor response rate and durability of response. An improvement in survival or disease-related symptoms has not yet been established. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
Merck is advancing a broad and fast-growing clinical development program for KEYTRUDA with more than 100 clinical trials - across more than 30 tumor types and over 16,000 patients - both as a monotherapy and in combination with other therapies.
Selected Important Safety Information for KEYTRUDA
Pneumonitis occurred in 12 (2.9%) of 411 patients with advanced melanoma receiving KEYTRUDA (the approved indication in the United States), including Grade 2 or 3 cases in 8 (1.9%) and 1 (0.2%) patients, respectively. Monitor patients for signs and symptoms of pneumonitis. Evaluate suspected pneumonitis with radiographic imaging. Administer corticosteroids for Grade 2 or greater pneumonitis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 pneumonitis.
Colitis (including microscopic colitis) occurred in 4 (1%) of 411 patients, including Grade 2 or 3 cases in 1 (0.2%) and 2 (0.5%) patients respectively, receiving KEYTRUDA. Monitor patients for signs and symptoms of colitis. Administer corticosteroids for Grade 2 or greater colitis. Withhold KEYTRUDA for Grade 2 or 3; permanently discontinue KEYTRUDA for Grade 4 colitis.
Hepatitis (including autoimmune hepatitis) occurred in 2 (0.5%) of 411 patients, including a Grade 4 case in 1 (0.2%) patient, receiving KEYTRUDA. Monitor patients for changes in liver function. Administer corticosteroids for Grade 2 or greater hepatitis and, based on severity of liver enzyme elevations, withhold or discontinue KEYTRUDA.
Hypophysitis occurred in 2 (0.5%) of 411 patients, including a Grade 2 case in 1 and a Grade 4 case in 1 (0.2% each) patient, receiving KEYTRUDA. Monitor for signs and symptoms of hypophysitis (including hypopituitarism and renal insufficiency). Administer corticosteroids for Grade 2 or greater hypophysitis. Withhold KEYTRUDA for Grade 2; withhold or discontinue for Grade 3; and permanently discontinue KEYTRUDA for Grade 4 hypophysitis.
Hyperthyroidism occurred in 5 (1.2%) of 411 patients, including Grade 2 or 3 cases in 2 (0.5%) and 1 (0.2%) patients, respectively, receiving KEYTRUDA. Hypothyroidism occurred in 34 (8.3%) of 411 patients, including a Grade 3 case in 1 (0.2%) patient, receiving KEYTRUDA. Thyroid disorders can occur at any time during treatment. Monitor patients for changes in thyroid function (at the start of treatment, periodically during treatment, and as indicated based on clinical evaluation) and for clinical signs and symptoms of thyroid disorders. Administer corticosteroids for Grade 3 or greater hyperthyroidism. Withhold KEYTRUDA for Grade 3; permanently discontinue KEYTRUDA for Grade 4 hyperthyroidism. Isolated hypothyroidism may be managed with replacement therapy without treatment interruption and without corticosteroids.
Type 1 diabetes mellitus, including diabetic ketoacidosis, has occurred in patients receiving KEYTRUDA. Monitor patients for hyperglycemia and other signs and symptoms of diabetes. Administer insulin for type 1 diabetes, and withhold KEYTRUDA in cases of severe hyperglycemia until metabolic control is achieved.
Nephritis occurred in 3 (0.7%) patients receiving KEYTRUDA, consisting of one case of Grade 2 autoimmune nephritis (0.2%) and two cases of interstitial nephritis with renal failure (0.5%), one Grade 3 and one Grade 4. Monitor patients for changes in renal function. Administer corticosteroids for Grade 2 or greater nephritis. Withhold KEYTRUDA for Grade 2; permanently discontinue KEYTRUDA for Grade 3 or 4 nephritis.
Other clinically important immune-mediated adverse reactions can occur. The following clinically significant, immune-mediated adverse reactions occurred in patients treated with KEYTRUDA: exfoliative dermatitis, uveitis, arthritis, myositis, pancreatitis, hemolytic anemia, partial seizures arising in a patient with inflammatory foci in brain parenchyma, severe dermatitis including bullous pemphigoid, myasthenic syndrome, optic neuritis, and rhabdomyolysis.
For suspected immune-mediated adverse reactions, ensure adequate evaluation to confirm etiology or exclude other causes. Based on the severity of the adverse reaction, withhold KEYTRUDA and administer corticosteroids. Upon improvement of the adverse reaction to Grade 1 or less, initiate corticosteroid taper and continue to taper over at least 1 month. Restart KEYTRUDA if the adverse reaction remains at Grade 1 or less. Permanently discontinue KEYTRUDA for any severe or Grade 3 immune-mediated adverse reaction that recurs and for any life-threatening immune-mediated adverse reaction.
Infusion-related reactions, including severe and life-threatening reactions, have occurred in patients receiving KEYTRUDA. Monitor patients for signs and symptoms of infusion-related reactions including rigors, chills, wheezing, pruritis, flushing, rash, hypotension, hypoxemia, and fever. For severe or life-threatening reactions, stop infusion and permanently discontinue KEYTRUDA.
Based on its mechanism of action, KEYTRUDA may cause fetal harm when administered to a pregnant woman. If used during pregnancy, or if the patient becomes pregnant during treatment, apprise the patient of the potential hazard to a fetus. Advise females of reproductive potential to use highly effective contraception during treatment and for 4 months after the last dose of KEYTRUDA.
For the treatment of advanced melanoma, KEYTRUDA was discontinued for adverse reactions in 9% of 411 patients across all doses studied. Serious adverse reactions occurred in 36% of patients receiving KEYTRUDA. The most frequent serious adverse drug reactions reported in 2% or more of patients were renal failure, dyspnea, pneumonia, and cellulitis.
The most common adverse reactions (reported in at least 20% of patients) were fatigue (47%), cough (30%), nausea (30%), pruritus (30%), rash (29%), decreased appetite (26%), constipation (21%), arthralgia (20%), and diarrhea (20%).
The recommended dose of KEYTRUDA is 2 mg/kg administered as an intravenous infusion over 30 minutes every three weeks until disease progression or unacceptable toxicity. No formal pharmacokinetic drug interaction studies have been conducted with KEYTRUDA. It is not known whether KEYTRUDA is excreted in human milk. Because many drugs are excreted in human milk, instruct women to discontinue nursing during treatment with KEYTRUDA. Safety and effectiveness of KEYTRUDA have not been established in pediatric patients.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while also enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the primary focus of Immune Design's ongoing immuno-oncology clinical programs, are products of its two synergistic discovery platforms, ZVexTM and GLAASTM, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Forward-Looking Statement of Immune Design
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the timing of initiation, progress, scope and outcome of clinical trials for Immune Design's product candidates and the reporting of clinical data regarding Immune Design's product candidates. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, failure of Immune Design's collaborators to support or advance collaborations or product candidates and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
Merck's Focus on Cancer
Our goal is to translate breakthrough science into innovative oncology medicines to help people with cancer worldwide. At Merck Oncology, helping people fight cancer is our passion and supporting accessibility to our cancer medicines is our commitment. Our focus is on pursuing research in immuno-oncology and we are accelerating every step in the journey - from lab to clinic - to potentially bring new hope to people with cancer. For more information about our oncology clinical trials, visit www.merck.com/clinicaltrials.
About Merck
Today's Merck is a global healthcare leader working to help the world be well. Merck is known as MSD outside of the United States and Canada. Through our prescription medicines, vaccines, biologic therapies and animal health products, we work with customers and operate in more than 140 countries to deliver innovative health solutions. We also demonstrate our commitment to increasing access to healthcare through far-reaching policies, programs and partnerships. For more information, visit www.merck.com and connect with us on Twitter, Facebook and YouTube.
Forward-Looking Statement of Merck & Co., Inc., Kenilworth, N.J., USA
This news release of Merck & Co., Inc., Kenilworth, N.J., USA (the "company") includes "forward-looking statements" within the meaning of the safe harbor provisions of the U.S. Private Securities Litigation Reform Act of 1995. These statements are based upon the current beliefs and expectations of the company's management and are subject to significant risks and uncertainties. There can be no guarantees with respect to pipeline products that the products will receive the necessary regulatory approvals or that they will prove to be commercially successful. If underlying assumptions prove inaccurate or risks or uncertainties materialize, actual results may differ materially from those set forth in the forward-looking statements.
Risks and uncertainties include but are not limited to, general industry conditions and competition; general economic factors, including interest rate and currency exchange rate fluctuations; the impact of pharmaceutical industry regulation and health care legislation in the United States and internationally; global trends toward health care cost containment; technological advances, new products and patents attained by competitors; challenges inherent in new product development, including obtaining regulatory approval; the company's ability to accurately predict future market conditions; manufacturing difficulties or delays; financial instability of international economies and sovereign risk; dependence on the effectiveness of the company's patents and other protections for innovative products; and the exposure to litigation, including patent litigation, and/or regulatory actions.
The company undertakes no obligation to publicly update any forward-looking statement, whether as a result of new information, future events or otherwise. Additional factors that could cause results to differ materially from those described in the forward-looking statements can be found in the company's 2014 Annual Report on Form 10-K and the company's other filings with the Securities and Exchange Commission (SEC) available at the SEC's Internet site (www.sec.gov).
Please see Prescribing Information for KEYTRUDA (pembrolizumab) at
http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_pi.pdf and the Medication Guide for
KEYTRUDA at http://www.merck.com/product/usa/pi_circulars/k/keytruda/keytruda_mg.pdf
CONTACT:
Immune Design
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
Merck
Media Contact
Pamela Eisele
(267) 305-3558
Investor Contact
Justin Holko
(908) 740-1879
GAITHERSBURG, Md.--(BUSINESS WIRE)-- GlycoMimetics, Inc. (NASDAQ: GLYC) announced today that Pfizer Inc. (NYSE: PFE) has dosed the first patient in the RESET (Rivipansel: Evaluating Safety, Efficacy and Time to Discharge) study - a Phase 3 clinical trial assessing the efficacy and safety of rivipansel for the treatment of vaso-occlusive crisis (VOC) in patients hospitalized with sickle cell disease who are six years of age or older. The start of this trial triggered the second of two milestone payments from Pfizer to GlycoMimetics totaling $35 million for Phase 3 initiation. GlycoMimetics received a $15 million milestone payment from Pfizer in May 2014.
According to Rachel King, Chief Executive Officer of GlycoMimetics, "The initiation of the Phase 3 trial is important progress toward our vision for an effective therapy for people experiencing sickle cell crisis. It's rewarding for the GlycoMimetics team to see this milestone reached."
This Phase 3, multicenter, randomized, double-blind, placebo-controlled, parallel-group study is planning to enroll at least 350 individuals with sickle cell disease, aged six and older who are hospitalized for a vaso-occlusive crisis, and will evaluate the efficacy and safety of treatment with rivipansel. Trial participants must be receiving treatment with intravenous opioids for their vaso-occlusive crisis and must be able to receive the first dose of study drug within 24 hours of initiation of intravenous opioid therapy. The primary endpoint for the study will be time to readiness-for-discharge. Key secondary endpoints will include time to discharge, cumulative IV opioid consumption and time to discontinuation of IV opioids. For additional information about the RESET Trial and to learn more about eligibility, patients can visit www.resetsicklecell.com.
In July 2014, GlycoMimetics announced that Pfizer had reached agreement with the U.S. Food & Drug Administration (FDA), under a special protocol assessment (SPA), for the Phase 3 clinical trial of rivipansel. The SPA serves as an agreement between Pfizer and the FDA regarding the design, endpoints and statistical analysis approach of a Phase 3 clinical trial, results from which could potentially support approval of a New Drug Application (NDA). This includes specific agreement on the approvable composite primary endpoint, time to readiness-for-discharge, and the key secondary endpoints (time to discharge, cumulative IV opioid consumption, and time to discontinuation of IV opioids) considered supportive but not sufficient for approval individually.
GlycoMimetics reported top line data from the Phase 2 trial of rivipansel in April 2013 and presented full data from the clinical trial in two oral presentations and one poster presentation at the December 2013 meeting of the American Society of Hematology (ASH). The oral presentations were selected as "Best of ASH." In the Phase 2 trial, patients treated with rivipansel experienced meaningful reductions in time to reach resolution of VOC, length of hospital stay and use of opioid analgesics for pain management, in each case as compared to patients receiving placebo.
In 2011, Pfizer and GlycoMimetics entered into a worldwide license agreement for the development and, if approved by applicable regulatory authorities, commercialization of rivipansel. GlycoMimetics was responsible for development through the Phase 2 clinical trial and Pfizer is now responsible for all future clinical development of rivipansel.
Rivipansel has previously received both Orphan Drug and Fast Track status for the treatment of VOC from the FDA, and Orphan Product status in the European Union.
About Sickle Cell Disease and VOC
Sickle cell disease is a genetic disease affecting 90,000 to 100,000 people in the United States, predominantly of African descent. One of the most severe complications of sickle cell disease is vaso-occlusive crisis (VOC). VOC is typically characterized by excruciating, debilitating pain that occurs periodically throughout the life of a person with sickle cell disease. VOC is responsible for more than 73,000 hospitalizations per year in the United States with an average hospital stay of approximately six days. The current standard of care for VOC consists of supportive therapy, primarily in the form of hydration and pain management, typically requiring extended hospitalization.
About GlycoMimetics, Inc.
GlycoMimetics is a clinical stage biotechnology company focused on the discovery and development of novel glycomimetic drugs to address unmet medical needs resulting from diseases in which carbohydrate biology plays a key role. Pfizer is the company's development partner for rivipansel, a GlycoMimetics-discovered investigational therapy for pain crisis associated with sickle cell disease, and is conducting a Phase 3 clinical study. A GlycoMimetics wholly-owned candidate therapy (GMI- 1271) for acute myeloid leukemia (AML) and other blood disorders is also in clinical trials. Glycomimetics are molecules that mimic the structure of carbohydrates involved in important biological processes. Using its expertise in carbohydrate chemistry and knowledge of carbohydrate biology, GlycoMimetics is developing a pipeline of glycomimetic drug candidates that inhibit disease-related functions of carbohydrates, such as the roles they play in inflammation, cancer and infection. Learn more at www.glycomimetics.com.
Cautionary Note Regarding Forward-Looking Statements
This press release contains forward-looking statements regarding the clinical development of rivipansel, including the recently initiated Phase 3 clinical trial. Actual results may differ materially from those in these forward-looking statements. For a further description of the risks associated with these statements, as well as other risks facing GlycoMimetics, please see the risk factors described in the Company's annual report on Form 10-K that was filed with the U.S. Securities and Exchange Commission on March 16, 2015, and other filings the Company makes with the SEC from time to time. Forward-looking statements speak only as of the date of this release, and GlycoMimetics undertakes no obligation to update or revise these statements, except as may be required by law.
View source version on businesswire.com: http://www.businesswire.com/news/home/20150623006403/en/
GlycoMimetics, Inc.
Investor Contact:
Shari Annes, 650-888-0902
sannes@annesassociates.com
or
Media Contact:
Jamie Lacey-Moreira, 410-299-3310
jamielacey@presscommpr.com
Source: GlycoMimetics
News Provided by Acquire Media
- Proceeds from the oversubscribed funding round will support the expansion of Unum’s proprietary pipeline and development of ACTR therapies for a broad range of hematological and solid tumor indications -
CAMBRIDGE, MA – June 11, 2015 –Unum Therapeutics, a biotechnology company developing a universal, antibody-directed cellular immunotherapy, announced today the close of a Series B financing totaling $65 million. New investor New Leaf Venture Partners led the round, with participation from additional new investors Brace Pharma Capital, Cowen Private Investments, Jennison Associates (on behalf of certain clients), Novo A/S, Sabby Management LLC, Sectoral Asset Management, and Wellington Management Company LLP. Unum’s existing investors – Fidelity Biosciences, Atlas Venture and Sanofi-Genzyme BioVentures – also participated in the oversubscribed round. In addition, Seattle Genetics, Inc. (Nasdaq: SGEN) made a $5 million equity investment in this round as part of the strategic collaboration between the companies announced earlier this week.
Liam Ratcliffe, M.D., Ph.D., Managing Director at New Leaf Venture Partners, who has been appointed to the Unum Board of Directors, stated, “We are thrilled to lead this financing into Unum. The ability to combine T cell therapy with a range of antibodies provides Unum a truly novel platform with numerous advantages, including rapidly developing cell therapies against multiple targets. The company is led by a world-class team and, together with the strength of this syndicate, is well-positioned to translate this potential into reality for cancer patients, and all stakeholders.”
The proceeds from the financing will be used to advance the Unum’s novel Antibody-Coupled T-cell Receptor (ACTR) technology platform and support the expansion of the company’s proprietary pipeline of ACTR therapies targeting a broad range of hematological and solid tumor targets.
“This is a unique time in the battle to develop cancer therapies. Across the industry, we’re seeing truly profound patient responses achieved by directing the immune system to attack tumor cells. This Series B funding will be critical for Unum, enabling us to advance our engineered T-cell therapy through clinical proof-of-concept and to simultaneously create a pipeline of programs targeting a range of cancer indications,” said Charles Wilson, Ph.D., President and Chief Executive Officer of Unum Therapeutics. “We are excited at the opportunity to use our ACTR cell therapy to address the unmet medical needs of patients with hematologic or solid tumor cancers.”
About ACTR Technology
ACTR is a chimeric protein that combines components from receptors normally found on two different human immune cell types – natural killer (NK) cells and T-cells – to create a novel cancer cell killing activity. T-cells bearing the ACTR receptor can be directed to attack tumor cells by providing a monoclonal antibody that binds to antigens on the cancer cell surface and then acts as a bridge to the ACTR T-cell, enabling tumor cell killing. Unum has built a platform for cancer treatment based upon ACTR. In contrast to other approaches that are limited to a single target and treat a narrow set of tumors, Unum’s approach is not restricted by antigen and may have applications for treating many types of cancers.
About Unum Therapeutics
Unum Therapeutics uses proprietary T-cell engineering technology in combination with tumor-targeting antibodies to activate the body’s own immune system to fight cancer. Unum’s lead program, based on its Antibody-Coupled T-cell Receptor (ACTR) technology, recently entered Phase 1 clinical testing to assess safety and efficacy. The company is headquartered in Cambridge, MA. For more information, visit www.unumrx.com.
-Collaboration Combines Seattle Genetics’ Expertise in Cancer Targets and Antibody-Based Therapies with Unum’s Novel Antibody-Coupled T-cell Receptor (ACTR) Technology-
-Companies to Focus on the Development of Next Generation Cellular Immunotherapy Agents that Combine Unum’s Universal T-cell Approach with Select Seattle Genetics Targets and Antibodies-
BOTHELL, WA and CAMBRIDGE, MA – June 8, 2015 – Seattle Genetics, Inc. (Nasdaq: SGEN) and Unum Therapeutics announced today that the two companies have entered into a strategic collaboration and license agreement to develop and commercialize novel antibody-coupled T-cell receptor (ACTR) therapies for cancer.
Unum’s proprietary ACTR technology enables programming of a patient’s T-cells to attack tumor cells when co-administered with tumor-specific therapeutic antibodies. Seattle Genetics, through its extensive work in the field of antibody-drug conjugates (ADCs), has a substantial portfolio of cancer targets and tumor-specific monoclonal antibodies from which programs will be selected for the collaboration.
“This collaboration is an exciting extension of our work over more than 17 years, empowering antibodies in order to provide new therapeutic options for cancer patients,” said Clay B. Siegall, Ph.D., President and Chief Executive Officer of Seattle Genetics. “Unum’s innovative technology for a universal, antibody-directed cellular immunotherapy is differentiated from other engineered Tcell approaches, and may have broad applicability across a range of cancer targets. We are pleased to be collaborating with one of the most promising companies in the emerging field of cellular immunotherapy to develop new treatment options for cancer patients with unmet medical needs.”
“Unum’s strategy is to develop and commercialize a universal cellular immunotherapy that can be used in combination with a variety of antibodies to attack a wide range of hematological and solid tumors,” said Charles Wilson, Ph.D., President and Chief Executive Officer of Unum Therapeutics. “We believe that our unique approach has the potential to advance beyond the safety and efficacy limitations of current generation T-cell approaches. We are delighted to collaborate with Seattle Genetics in the development of ACTR therapies. Their leadership in antibody-based therapies and expertise in the development of cancer treatments will be invaluable as we work together to bring potentially breakthrough therapies to patients.”
Under the terms of the agreement, Seattle Genetics will make an upfront payment of $25 million and an equity investment of $5 million in Unum’s next round of private financing. The companies will initially develop two ACTR products incorporating Seattle Genetics’ antibodies, and Seattle Genetics has an option to expand the collaboration to include a third ACTR product. Unum will conduct preclinical research and clinical development activities through phase 1 with funding from Seattle Genetics. The companies will work together to co-develop and jointly fund programs after phase 1 unless either company opts out. Seattle Genetics and Unum will co-commercialize and share profits 50/50 on any co-developed programs in the United States. Seattle Genetics will retain exclusive commercial rights outside of the United States, paying Unum high single to mid-double digit royalties on ex-U.S. sales. Potential option fee and progress-dependent milestone payments to Unum under the collaboration may total up to $615 million across all three ACTR programs.
As a result of the amounts paid up front and the additional development activities expected under this deal, Seattle Genetics will provide revised 2015 financial guidance in connection with announcing its second quarter financial results currently planned for July 30, 2015.
About ACTR Technology
ACTR is a chimeric protein that combines components from receptors normally found on two different human immune cell types – natural killer (NK) cells and T-cells – to create a novel cancer cell killing activity. T-cells bearing the ACTR receptor can be directed to attack tumor cells by providing a monoclonal antibody that binds to antigens on the cancer cell surface and then acts as a bridge to the ACTR T-cell, enabling tumor cell killing. Unum has built a platform for cancer treatment based upon ACTR. In contrast to other approaches that are limited to a single target and treat a narrow set of tumors, Unum’s approach is not restricted by antigen and may have applications
for treating many types of cancers.
About Seattle Genetics
Seattle Genetics is a biotechnology company focused on the development and commercialization of innovative antibody-based therapies for the treatment of cancer. Seattle Genetics is leading the field in developing antibody-drug conjugates (ADCs), a technology designed to harness the targeting ability of antibodies to deliver cell-killing agents directly to cancer cells. The company’s lead product, ADCETRIS® (brentuximab vedotin) is a CD30-targeted ADC that, in collaboration with Takeda Pharmaceutical Company Limited, is commercially available for two indications in more than 55 countries, including the U.S., Canada, Japan and members of the European Union.
Additionally, ADCETRIS is being evaluated broadly in more than 30 ongoing clinical trials in CD30-expressing malignancies. Seattle Genetics is also advancing a robust pipeline of clinical-stage programs, including SGN-CD19A, SGN-CD33A, SGN-LIV1A, SGN-CD70A, ASG-22ME, ASG- 15ME and SEA-CD40. Seattle Genetics has collaborations for its ADC technology with a number of leading biotechnology and pharmaceutical companies, including AbbVie, Agensys (an affiliate of Astellas), Bayer, Genentech, GlaxoSmithKline and Pfizer. More information can be found at www.seattlegenetics.com.
About Unum Therapeutics
Unum Therapeutics uses proprietary T-cell engineering technology in combination with tumortargeting antibodies to activate the body’s own immune system to fight cancer. Unum’s lead program, based on its Antibody-Coupled T-cell Receptor (ACTR) technology, recently entered Phase 1 clinical testing to assess safety and efficacy. Unum is seeking partners interested in using the ACTR technology to arm proprietary tumor-specific antibodies with a T-cell to improve their therapeutic potential. The company is headquartered in Cambridge, MA. For more information, visit
www.unumrx.com.
For Seattle Genetics:
Certain of the statements made in this press release are forward looking, such as those, among others, relating to the therapeutic potential of ACTR-based products. Actual results or developments may differ materially from those projected or implied in these forward-looking statements. Factors that may cause such a difference include the inability to show sufficient activity in clinical trials and the risk of adverse events as these programs advance in clinical trials. More information about the risks and uncertainties faced by Seattle Genetics is contained in the company’s 10-Q for the quarter ended March 31, 2015 filed with the Securities and Exchange Commission. Seattle Genetics disclaims any intention or obligation to update or revise any forward-looking statements, whether as a result of new information, future events or otherwise.
# # #
CONTACTS
Seattle Genetics:
Investors:
Peggy Pinkston
(425) 527-4160
ppinkston@seagen.com
Media:
Tricia Larson
(425) 527-4180
tlarson@seagen.com
Unum Therapeutics:
Mariesa Kemble
Sam Brown Inc.
(608) 850-4745
mariesakemble@sambrown.com

Proof-of-Concept Data in Well-Established Model of Type 1 Diabetes Presented Today at the 75th Scientific Sessions of the American Diabetes Association
SAN DIEGO, June 5, 2015 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE) announced today that it has entered into a two-year sponsored research agreement with Boston Children's Hospital to accelerate the development of an adoptive immunoregulatory cell therapy to treat autoimmune diseases. The collaboration seeks to assess the potential of Fate's PD-L1 programmed CD34+ cellular therapeutic as a transformative treatment for type 1 diabetes. The Company's adoptive immunoregulatory cell therapy is currently undergoing preclinical testing across a range of autoimmune and inflammatory diseases.
The research program will be led by Paolo Fiorina, M.D., Ph.D., Assistant Professor of Pediatrics at Boston Children's Hospital and Harvard Medical School. Under the agreement, Dr. Fiorina will investigate the potential of Fate's PD-L1 programmed CD34+ cellular therapeutic to abrogate autoimmune activity responsible for the destruction of pancreatic beta cells and the development of type 1 diabetes. Preclinical data from the Fiorina laboratory presented today at the American Diabetes Association's 75th Scientific Sessions in Boston, Mass., shows that genetically engineered PD-L1+ hematopoietic cells adoptively transferred into hyperglycemic mice traffic to the pancreas, reduce aberrant T cell activity and revert hyperglycemia in a well-established murine model of type 1 diabetes.
"Our research revealed that human CD34+ cells from individuals with type 1 diabetes have reduced expression levels of PDL1, and that genetically engineered hematopoietic cells can induce anergy of auto-reactive T cells in vivo by leveraging the immunosuppressive properties of the PD-L1 / PD-1 pathway," said Dr. Fiorina. "Fate's clinically validated ex vivo cell programming platform offers a promising approach to therapeutically harness these exciting preclinical proof-of-concept data, and we look forward to evaluating the potential of the Company's adoptive immunoregulatory cell therapy to transform the treatment of type 1 diabetes."
Dr. Fiorina and his team have extensively studied the cellular mechanisms and molecular pathways involved in the autoimmune-mediated destruction of pancreatic beta cells that result in insulin deficiency and onset of type 1 diabetes. While life-long daily insulin treatment allows for chronic management of type 1 diabetes, there remains a large unmet need for disease-modifying treatments that directly address the autoimmune etiology of the disease. Each year, more than 15,000 children and 15,000 adults —approximately 80 people per day —are diagnosed with type 1 diabetes in the US alone. Adoptive immunoregulatory cell therapy represents a novel approach to restoring immune homeostasis and inducing long-term tolerance in patients with type 1 diabetes and other autoimmune disorders.
The partnership brings together Boston Children's pioneering research on the immunoregulatory properties of hematopoietic cells and Fate's innovative platform for programming the in vivo biological activity and therapeutic potential of hematopoietic cellular therapeutics. Using its platform, Fate Therapeutics discovered a combination of pharmacologic modulators to promote rapid and supra-physiologic activation of PD-L1 on the surface of human CD34+ hematopoietic cells. The Company has shown that these programmed CD34+ cells recognize and significantly reduce the proliferation rates of activated T cells in vitro as compared to unmodulated human CD34+ hematopoietic cells.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the development of programmed cellular therapeutics for the treatment of severe, life-threatening diseases. The Company's approach utilizes established pharmacologic modalities, such as small molecules, to program the fate and function of cells ex vivo. The Company's lead product candidate, PROHEMA®, is an ex vivo programmed hematopoietic cellular therapeutic, which is currently in clinical development for the treatment of hematologic malignancies and rare genetic disorders in patients undergoing hematopoietic stem cell transplantation (HSCT). The Company is also using its proprietary induced pluripotent stem cell platform to develop ex vivo reprogrammed hematopoietic and myogenic cellular therapeutics. Fate Therapeutics is headquartered in San Diego, CA. For more information, please visit www.fatetherapeutics.com.
Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the therapeutic potential of pharmacologically modulated CD34+ hematopoietic cells and any product candidates that may arise from the Company's collaboration with Boston Children's Hospital, and the Company's plans to undertake certain preclinical research and development of programmed hematopoietic cells. These and any other forwardlooking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, the risk of cessation or delay of planned preclinical development activities for a variety of reasons, any inability to develop programmed CD34+ hematopoietic cells suitable for therapeutic applications, the risk that programmed human CD34+ hematopoietic cells may not produce the therapeutic benefits suggested by the results observed in preclinical studies, and the risk that the Company's collaboration with Boston Children's Hospital may not be successful or may be terminated for a variety of reasons. For a discussion of other risks and uncertainties, and other important factors, any of which could cause our actual results to differ from those contained in the forward-looking statements, see the risks and uncertainties detailed in the Company's periodic filings with the Securities and Exchange Commission, including but not limited to the Company's Form 10-Q for the quarter ended March 31, 2015, and from time to time the Company's other investor communications. We are providing the information in this release as of this date and do not undertake any obligation to update any forward-looking statements contained in this release as a result of new information, future events or otherwise.
CONTACT:
Jesse Baumgartner, Stern Investor Relations, Inc.
212.362.1200, jesse@sternir.com
Source: Fate Therapeutics, Inc
News Provided by Acquire Media
SAN DIEGO, May 20, 2015 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE), a biopharmaceutical company engaged in the development of programmed cellular therapeutics for the treatment of severe, life-threatening diseases, today announced the pricing of an underwritten public offering of 6,000,000 shares of its common stock at a public offering price of $5.00 per share, before underwriting discounts, for an aggregate offering of $30 million. Fate Therapeutics has granted the underwriters a 30-day option to purchase up to an additional 900,000 shares of its common stock. The proceeds to Fate Therapeutics from this offering are expected to be approximately $27.7 million after deducting underwriting discounts and commissions and other estimated offering expenses but excluding any exercise of the underwriters' option. Fate Therapeutics intends to use the net proceeds from the offering for clinical development and research activities, working capital and other general corporate purposes. All shares of common stock to be sold in the offering are being offered by Fate Therapeutics. The offering is expected to close on or about May 27, 2015, subject to customary closing conditions.
Leerink Partners LLC and BMO Capital Markets Corp. are acting as joint book-running managers for the offering. Wedbush PacGrow and H.C. Wainwright & Co., LLC are acting as co-managers.
The securities described above are being offered by Fate Therapeutics pursuant to a shelf registration statement on Form S-3 (File No. 333-199107) previously filed with and declared effective by the Securities and Exchange Commission (the "SEC"). The securities may be offered only by means of a prospectus. A preliminary prospectus supplement related to the offering was filed with the SEC on May 18, 2015 and is available on the SEC's website at http://www.sec.gov and a final prospectus supplement related to the offering will be filed with the SEC and will be available on the SEC's website at http://www.sec.gov. Copies of the final prospectus supplement and the accompanying prospectus for the securities being offered may also be obtained, when available, from Leerink Partners LLC, Attention: Syndicate Department, One Federal Street, 37th Floor, Boston, MA 02110, or by email at syndicate@leerink.com, or by phone at (800) 808‐7525, ext. 6142; or from BMO Capital Markets Corp. at 3 Times Square, 26th Floor, New York, NY 10036, Attention: Equity Syndicate Department, by telephone at (800) 414-3627 or by email to bmoprospectus@bmo.com.
This press release does not constitute an offer to sell or the solicitation of an offer to buy, nor shall there be any sale of these securities in any state or jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the development of programmed cellular therapeutics for the treatment of severe, life-threatening diseases. The Company's approach utilizes established pharmacologic modalities, such as small molecules, to program the fate and function of cells ex vivo. The Company's lead product candidate, PROHEMA®, is an ex vivo programmed hematopoietic cellular therapeutic, which is currently in clinical development for the treatment of hematologic malignancies and rare genetic disorders in patients undergoing hematopoietic stem cell transplantation (HSCT). The Company is also using its proprietary induced pluripotent stem cell platform to develop ex vivo reprogrammed hematopoietic and myogenic cellular therapeutics. Fate Therapeutics is headquartered in San Diego, CA.
Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding Fate Therapeutics' expectations with respect to the offering described in this press release, including its ability to complete the offering and its expected and intended use of proceeds from the offering. These and any other forward-looking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, the risks and uncertainties associated with market conditions and the satisfaction of customary closing conditions related to the proposed offering, as well as risks and uncertainties detailed in the Company's periodic filings with the Securities and Exchange Commission, including but not limited to the Company's Form 10-Q for the quarter ended March 31, 2015, and from time to time the Company's other investor communications. Fate Therapeutics is providing the information in this release as of this date and does not undertake any obligation to update any forward-looking statements contained in this release as a result of new information, future events or otherwise, except to the extent required by law.
CONTACT:
Jesse Baumgartner, Stern Investor Relations, Inc.
212.362.1200, jesse@sternir.com
Source: Fate Therapeutics, Inc
News Provided by Acquire Media
SAN DIEGO, May 18, 2015 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE), a biopharmaceutical company engaged in the development of programmed cellular therapeutics for the treatment of severe, life-threatening diseases, today announced that it has commenced an underwritten public offering of 6,000,000 shares of its common stock. Fate Therapeutics intends to grant the underwriters a 30-day option to purchase up to an additional 900,000 shares of its common stock. Fate Therapeutics intends to use the net proceeds from the offering for clinical development and research activities, working capital and other general corporate purposes. All shares of common stock to be sold in the offering will be offered by Fate Therapeutics. The offering is subject to market conditions, and there can be no assurance as to whether or when the offering may be completed, or the actual size or terms of the offering.
Leerink Partners LLC and BMO Capital Markets Corp. are acting as joint book-running managers for the offering. Wedbush PacGrow and H.C. Wainwright & Co., LLC are acting as co-managers.
The securities described above are being offered by Fate Therapeutics pursuant to a shelf registration statement on Form S-3 (File No. 333-199107) previously filed with and declared effective by the Securities and Exchange Commission (the "SEC").
A preliminary prospectus supplement and accompanying prospectus relating to the offering will be filed with the SEC and will be available on the SEC's website at http://www.sec.gov. A copy of the preliminary prospectus and accompanying prospectus can be obtained from Leerink Partners LLC, Attention: Syndicate Department, One Federal Street, 37th Floor, Boston, MA 02110, or by email at syndicate@leerink.com, or by phone at (800) 808-7525, ext. 6142; or from BMO Capital Markets Corp. at 3 Times Square, 26th Floor, New York, NY 10036, Attention: Equity Syndicate Department, by telephone at (800) 414-3627 or by email to bmoprospectus@bmo.com.
This press release does not constitute an offer to sell or the solicitation of an offer to buy these securities, nor shall there be any sale of these securities in any state or jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the development of programmed cellular therapeutics for the treatment of severe, life-threatening diseases. The Company's approach utilizes established pharmacologic modalities, such as small molecules, to program the fate and function of cells ex vivo. The Company's lead product candidate, PROHEMA®, is an ex vivo programmed hematopoietic cellular therapeutic, which is currently in clinical development for the treatment of hematologic malignancies and rare genetic disorders in patients undergoing hematopoietic stem cell transplantation (HSCT). The Company is also using its proprietary induced pluripotent stem cell platform to develop ex vivo reprogrammed hematopoietic and myogenic cellular therapeutics. Fate Therapeutics is headquartered in San Diego, CA.
Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding Fate Therapeutics' expectations with respect to its proposed offering, its intention to grant the underwriters an option to purchase additional shares and its intended use of proceeds from the offering. These and any other forward-looking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, the risks and uncertainties associated with market conditions and the satisfaction of customary closing conditions related to the proposed offering, as well as risks and uncertainties detailed in the Company's periodic filings with the Securities and Exchange Commission, including but not limited to the Company's Form 10-Q for the quarter ended March 31, 2015, and from time to time the Company's other investor communications. Fate Therapeutics is providing the information in this release as of this date and does not undertake any obligation to update any forward-looking statements contained in this release as a result of new information, future events or otherwise, except to the extent required by law.
CONTACT:
Jesse Baumgartner, Stern Investor Relations, Inc.
212.362.1200, jesse@sternir.com
Source: Fate Therapeutics, Inc
News Provided by Acquire Media
Alliance Utilizes Fate's Hematopoietic Cell Programming Platform to Identify Small Molecule Modulators
for Juno's Leading Genetically-Engineered T Cell Immunotherapies
SEATTLE and SAN DIEGO, May 6, 2015 (GLOBE NEWSWIRE) -- Juno Therapeutics, Inc. (Nasdaq:JUNO) and Fate Therapeutics, Inc. (Nasdaq:FATE) announced today that they have executed a strategic research collaboration and license agreement to identify and utilize small molecules to modulate Juno's genetically-engineered T cell product candidates to improve their therapeutic potential for cancer patients. The collaboration brings together Juno's industry-leading expertise in the development of chimeric antigen receptor (CAR) and T cell receptor (TCR) based cellular immunotherapies and Fate's innovative platform for programming the biological properties and in vivo therapeutic potential of hematopoietic cells.
"A deep understanding of T cell biology is the basis of Juno's approach to creating best-in-class cellular immunotherapies," said Hans Bishop, Chief Executive Officer of Juno Therapeutics. "Partnering with Fate Therapeutics, and accessing its strong science and leading platform for modulating the properties of immunological cells, enables interrogation of new avenues of T cell manipulation and provides an opportunity to enhance the therapeutic profile of our genetically-engineered T cell product candidates."
Through the four-year research and development collaboration, Fate will be responsible for screening and identifying small molecules that modulate the biological properties of engineered T cells. Juno will be responsible for the development and commercialization of engineered T cell immunotherapies incorporating Fate's small molecule modulators. Juno has the option to extend the exclusive research term for two years through an additional payment and continued funding of collaboration activities.
"We are excited to establish this strategic alliance with Juno, a company that shares our deep commitment to developing transformative cellular therapeutics for patients afflicted with life-threatening disorders," said Christian Weyer, M.D., M.A.S., President and Chief Executive Officer of Fate Therapeutics. "This partnership exemplifies the extension of our small molecule programming platform to additional hematopoietic cell types, such as T cells, as we continue to build and advance our innovative pipeline of programmed hematopoietic cellular therapeutic candidates."
Financial terms of the agreement include an upfront payment to Fate of $5 million and the purchase by Juno of one million shares of Fate common stock at $8.00 per share. Juno will fund all mutual collaboration activities for an exclusive four-year research term. For each product developed by Juno that incorporates modulators identified through the collaboration, Fate is eligible to receive approximately $50 million in target selection fees and clinical, regulatory and commercial milestones, as well as low single-digit royalties on sales. Fate retains exclusive rights to its intellectual property for all purposes outside of programmed CAR and TCR immunotherapies.
About Chimeric Antigen Receptor (CAR) Technology
Juno's chimeric antigen receptor (CAR) technology genetically engineers T cells to recognize and kill cancer cells. Juno's CAR T cell technology inserts a gene for a particular CAR into the T cell, enabling it to recognize cancer cells based on the expression of a specific protein located on the cell surface. When the engineered T cell engages the target protein on the cancer cell, it initiates a cell-killing response against the cancer cell.
About Cell Programming
Since its founding, Fate Therapeutics has been dedicated to programming the function of cells ex vivo to improve their therapeutic potential. Using advanced molecular characterization tools and technologies, Fate's platform enables the identification of small molecule or biologic modulators that promote rapid and supra-physiologic activation or inhibition of therapeutically-relevant genes and cell-surface proteins, such as those involved in the homing, proliferation and survival of hematopoietic stem cells or those involved in the persistence, proliferation and reactivity of immunological cells. Fate utilizes its deep understanding of the hematopoietic system to rapidly assess and quantify the therapeutic potential of programmed hematopoietic cells in vivo, and applies its modulators to maximize the safety and efficacy of hematopoietic cellular therapeutics.
About Juno Therapeutics, Inc.
Juno Therapeutics is building a fully integrated biopharmaceutical company focused on revolutionizing medicine by reengaging the body's immune system to treat cancer. Founded on the vision that the use of human cells as therapeutic entities will drive one of the next important phases in medicine, Juno is developing cell-based cancer immunotherapies based on chimeric antigen receptor and high-affinity T cell receptor technologies to genetically engineer T cells to recognize and kill cancer. Juno is developing multiple cell-based product candidates to treat a variety of B-cell malignancies as well as solid tumors. Several product candidates have shown compelling evidence of tumor shrinkage in the clinical trials in refractory leukemia and lymphoma conducted to date. Juno's long-term aim is to improve and leverage its cell-based platform to develop new product candidates that address a broader range of cancers and human diseases. Juno brings together innovative technologies from some of the world's leading research institutions, including the Fred Hutchinson Cancer Research Center, Memorial Sloan Kettering Cancer Center, Seattle Children's Research Institute, and The National Cancer Institute.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the development of programmed cellular therapeutics for the treatment of severe, life-threatening diseases. The Company's approach utilizes established pharmacologic modalities, such as small molecules, to program the fate and function of cells ex vivo. The Company's lead product candidate, PROHEMA®, is an ex vivo programmed hematopoietic cellular therapeutic, which is currently in clinical development for the treatment of hematologic malignancies and rare genetic disorders in patients undergoing hematopoietic stem cell transplantation (HSCT). The Company is also using its proprietary induced pluripotent stem cell platform to develop ex vivo reprogrammed hematopoietic and myogenic cellular therapeutics. Fate Therapeutics is headquartered in San Diego, CA. For more information, please visit www.fatetherapeutics.com.
Juno Forward Looking Statements
This press release contains forward-looking statements, including statements regarding the impact, benefits, timing, conduct, and funding of collaboration between Juno and Fate, as well as the capabilities, expertise, and responsibilities of each. Forward-looking statements are subject to risks and uncertainties that could cause actual results to differ materially from such forward-looking statements, and reported results should not be considered as an indication of future performance. These risks and uncertainties include, but are not limited to, risks associated with: the success, cost, and timing of Juno's product development activities and clinical trials, and Juno's ability to finance these activities and trials; Juno's ability to obtain regulatory approval for and to commercialize its product candidates; Juno's ability to establish a commercially-viable manufacturing process and manufacturing infrastructure; regulatory requirements and regulatory developments; success of Juno's competitors with respect to competing treatments and technologies; Juno's dependence on third-party research institution collaborators and other contractors in Juno's research and development activities, including for the conduct of clinical trials and the manufacture of Juno's product candidates; Juno's ability to obtain, maintain, or protect intellectual property rights related to its product candidates; amongst others. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to Juno's business in general, see Juno's Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 19, 2015 and Juno's other periodic reports filed with the Securities and Exchange Commission. These forward-looking statements speak only as of the date hereof. Juno disclaims any obligation to update these forward-looking statements.
Fate Therapeutics Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the Company's ability to identify and evaluate small molecule modulators for the programming of T cells, the Company's plans to undertake certain preclinical research on the therapeutic potential of programmed T cells, our expectations regarding the clinical effectiveness and safety of programmed T cell therapeutics, including CAR and TCR products developed through the collaboration, and the potential benefits of the collaboration, including expected funding and payments to be received under the collaboration. These and any other forward-looking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, the risk that we are unable to conduct or complete activities required under the collaboration, the risk of cessation or delay of any development activities under the collaboration for a variety of reasons (including any difficulties or delays in identifying modulators for the programming of T cells, and any adverse effects or events or other negative results that may be observed in clinical development of any product candidates developed through the collaboration), and the risk that funding and payments received under the collaboration may be less than expected. For a discussion of other risks and uncertainties, and other important factors, any of which could cause our actual results to differ from those contained in the forward-looking statements, see the risks and uncertainties detailed in the Company's periodic filings with the Securities and Exchange Commission, including but not limited to the Company's Form 10-K for the year ended December 31st, 2014, and from time to time the Company's other investor communications. We are providing the information in this release as of this date and do not undertake any obligation to update any forward-looking statements contained in this release as a result of new information, future events or otherwise.
CONTACT:
Juno Therapeutics Contacts
Investor Relations:
David Walsey, W2O Group
dwalsey@w2ogroup.com, 858-617-0772
Media:
Julie Normart, W2O Group
jnormart@w2ogroup.com, 415-946-1087
Fate Therapeutics Contact
Jesse Baumgartner, Stern Investor Relations, Inc.
212.362.1200, jesse@sternir.com
Source: Fate Therapeutics, Inc; Juno Therapeutics, Inc.
News Provided by Acquire Media
SEATTLE and SOUTH SAN FRANCISCO, Calif., April 15, 2015 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ) today announced the pricing of an underwritten public offering of 3,000,000 shares of its common stock at a price to the public of $26.50 per share for total gross proceeds of approximately $79.5 million, before deducting underwriting discounts and commissions and estimated expenses. All of the shares of common stock are being offered by Immune Design. In addition, Immune Design has granted the underwriters a 30-day option to purchase up to 450,000 additional shares of common stock at the offering price, less underwriting discounts and commissions. The offering is expected to close on April 21, 2015, subject to customary closing conditions.
Jefferies LLC, Leerink Partners LLC and Cowen and Company, LLC are acting as joint book-running managers, and Wells Fargo Securities, LLC is acting as lead manager for the offering.
A registration statement relating to the common stock being offered by Immune Design was declared effective by the Securities and Exchange Commission on April 15, 2015. The offering is being made only by means of a final prospectus, which is part of the effective registration statement. Copies of the final prospectus, when available, may be obtained by contacting Jefferies LLC, Attention: Equity Syndicate Prospectus Department, 520 Madison Avenue, 12th Floor, New York, NY 10022, telephone: (877) 547-6340, e-mail: Prospectus_Department@Jefferies.com; Leerink Partners LLC, Attention: Syndicate Department, One Federal Street, 37th Floor, Boston, MA 02110, by telephone at (800) 808-7525 ext. 6142, or by email at syndicate@leerink.com; or Cowen and Company, LLC, c/o Broadridge Financial Services, 1155 Long Island Avenue, Edgewood, NY, 11717, Attention: Prospectus Department, telephone: (631) 274-2806, fax: (631) 254-7140.
This press release shall not constitute an offer to sell or the solicitation of an offer to buy, nor shall there be any sale of these securities in any state or jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the two-pronged focus of Immune Design's ongoing immuno-oncology clinical programs, are the product of its two synergistic discovery platforms: ZVex and GLAAS. Immune Design has offices in Seattle and South San Francisco.
CONTACT:
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
SEATTLE and SAN FRANCISCO, March 31, 2015 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on cancer, reported today that analyses of data from three ongoing Phase 1 studies support continued development of its two primary product candidates, CMB305 and G100.
CMB305 and G100 represent Immune Design's two distinct approaches to fighting cancer via the in vivo induction and/or expansion of anti-tumor CD8 T cells. CMB305 belongs within the Specific Antigen approach and is a "first-in-class" prime-boost therapy containing two potentially synergistic agents, LV305 and G305. CMB305 targets the tumor antigen, NY-ESO-1, which is expressed in a wide range of tumors. In contrast, G100 belongs within the Endogenous Antigen, or intra-tumoral immune activation, approach and relies on activating an anti-tumor immune response by its administration directly into the tumor. G100 is expected to directly activate dendritic and other antigen presenting cells near the tumor, which may enhance the function of pre-existing cytotoxic T lymphocytes (CTLs) and create an immune response against neo-antigens. Both are designed to work in vivo and provide a potential "off the shelf" therapy, in contrast to other, ex vivo T cell approaches.
CMB305
CMB305's "prime" and "boost" components, LV305 and G305, were well tolerated and demonstrated specific and selective immunogenicity in two parallel Phase 1 studies to meet the company's guidelines to progress CMB305 into development:
- After reviewing the safety data from each of the two studies, the DSMBs for each study voted that each agent was safe without dose-limiting toxicities;
- A significant subset of LV305 treated patients had NY-ESO-1-specific CD8 T cell responses that were generated or increased after therapy;
- A significant subset of G305 patients had a combination of NY-ESO-1-specific CD4 T cells and antibody responses that were generated or increased after therapy; and
- Clinical benefit in the form of stable disease was observed in a number of patients.
G100
- l e ongoing safety analysis demonstrates an acceptable profile alone or in combination with local radiation; and
- In addition to the initial complete response previous reported, we have observed additional evidence of clinical efficacy.
Abstracts for each of these three Phase 1 studies have been submitted for presentation at the American Society of Clinical Oncology (ASCO) Annual Meeting (May 29-June 2, 2015). If afforded the opportunity to present, it is the company's intent to work with the Principal Investigator for each product candidate to present a more complete data set at the Conference.
"We are very encouraged by the data produced thus far in all three Phase 1 studies," said Carlos Paya, M.D., Ph.D, President and Chief Executive Officer of Immune Design. "The data from the dose escalation Phase 1 studies of LV305 and G305 met our internal requirements for demonstrating that each of these two 'building blocks' of CMB305 selectively activates different arms of the immune response against NY-ESO-1, thus providing a strong rationale for their combination into the unique 'primeboost' approach intended to generate in vivo anti-NY-ESO-1 CTLs in our recently announced Phase 1b trial of CMB305. Likewise, the continued evidence of the therapeutic effects of G100 alone or in combination with radiation further supports the development of G100 under the 'intra-tumoral immune activation' approach."
About CMB305
CMB305 is an immuno-oncology product candidate combining two potentially synergistic agents, LV305 and G305, and is part of Immune Design's Specific Antigen approach to treating cancer and is a product of the ZVexTM discovery platform. This approach is designed to deliver specific tumor antigens in RNA form directly to cancer patients' dendritic cells using a cuttingedge gene delivery vector specific for a subset of skin dendritic cells. CMB305 is designed to target NY-ESO-1-expressing cancers and is intended to be an "off-the shelf" therapy that does not require patient-specific manufacturing or ex vivo manipulation of patient samples. The NY-ESO-1 protein is provided by the Ludwig Institute for Cancer Research. CMB305 is currently being evaluated in a Phase 1b open label, dose escalation, multi-center trial designed to evaluate the safety and tolerability, immunogenicity, and preliminary clinical efficacy of CMB305 in patients with any of four solid tumor types that are locally advanced, relapsed or metastatic and express NY-ESO-1.
About G100
G100 is intended for intra-tumoral injection and is part of Immune Design's intra-tumoral Immune Activation/Endogenous Antigen approach to treating cancer, which leverages an intra-tumoral activation of dendritic cells in the tumor microenvironment to potentially create a robust local and systemic anti-tumor immune response. G100 is a product of the company's GLAASTM platform, and is currently being studied in two separate pilot Phase 1 studies: one in patients with Merkel Cell Carcinoma, or MCC; and one via an investigator-sponsored trial in sarcoma patients in combination with local radiation.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the two-pronged focus of Immune Design's ongoing immuno-oncology clinical programs, are the product of its two synergistic discovery platforms: ZVex and GLAAS, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the clinical development of Immune Design's product candidates. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, failure of Immune Design's collaborators to support or advance collaborations or product candidates and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
CONTACT:
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
Prime-Boost in vivo T cell active immunotherapy
SEATTLE and SOUTH SAN FRANCISCO, Calif., March 26, 2015 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company focused on cancer, today announced the dosing of patients in a Phase 1b clinical trial of CMB305, a "prime-boost" immuno-oncology product candidate generated from the company's ZVexTM and GLAASTM platforms. CMB305 is the first product candidate of Immune Design's Specific Antigen approach, which is one of the company's two distinct approaches to fighting cancer.
CMB305 is a novel combination product that involves the sequential dosing of two complementary agents, LV305 and G305, and is designed to synergistically induce anti-tumor cytotoxic T lymphocytes (CTLs) to target tumors that express NY-ESO-1, a tumor antigen found in a broad set of tumors.
"CMB305's prime-boost approach leverages the activation of two separate arms of the patient's immune system, that when combined under this novel approach, are designed to induce a strong and long lasting anti-tumor CD8 T cell response, as well as activate other complementary immune effectors that may help to fight cancer," said Carlos V. Paya, MD, PhD, chief executive officer of Immune Design.
The Phase 1b open label, multi-center trial (NCT02387125) is designed to evaluate the safety and tolerability, immunogenicity, and preliminary clinical efficacy of CMB305 in patients with locally advanced, relapsed or metastatic solid cancers expressing NY-ESO-1. The study is divided into two parts. Part 1 is a dose escalation study in up to 12 patients. Part 2 will be an expansion study of the optimal dose in approximately 27 patients.
About CMB305
CMB305 is an immuno-oncology product candidate combining two potentially synergistic agents. The first agent, LV305, is a hybrid vector from the ZVex platform that specifically targets dendritic cells (DCs) in vivo and delivers the RNA for NY-ESO-1, enabling the DCs to express the entire tumor antigen to potentially induce a diverse set of CTLs. G305 is the second agent and is designed to boost the CTL response via the induction of antigen-specific CD4 "helper" T cells. G305 consists of recombinant NY-ESO-1 protein formulated with a proprietary synthetic small molecule called glucopyranosyl lipid A (GLA), the novel TLR4 agonist at the core of the GLAAS platform. CMB305 is intended to be an "off-the shelf" therapy that does not require patient-specific manufacturing or ex vivo manipulation of patient samples. The NY-ESO-1 protein is provided by the Ludwig Institute for Cancer Research.
For Patients
The CMB305 trial is being conducted at a number of leading cancer research organizations. Information about this study, including additional eligibility criteria and contact information, can be found on clinicaltrials.gov.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while enhancing other immune effectors, to fight cancer and other chronic diseases. CMB305 and G100, the foci of Immune Design's on-going immuno-oncology clinical programs, are the product of its two synergistic discovery platforms: ZVex and GLAAS, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Cautionary Note on Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include, but are not limited to, statements about the expected mechanism and results of CMB-305 and its potential impact on cancer treatment. Many factors may cause differences between current expectations and actual results including unexpected safety or efficacy data observed during preclinical or clinical studies, clinical trial site activation or enrollment rates that are lower than expected, changes in expected or existing competition, changes in the regulatory environment, failure of Immune Design's collaborators to support or advance collaborations or product candidates and unexpected litigation or other disputes. Other factors that may cause Immune Design's actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" sections contained therein. Except as required by law, Immune Design assumes no obligation to update any forwardlooking statements contained herein to reflect any change in expectations, even as new information becomes available.
CONTACT:
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Shari Annes
Annes Associates
sannes@annesassociates.com
650-888-0902
SAN DIEGO, March 3, 2015 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE), a biopharmaceutical company engaged in the development of programmed cellular therapeutics for the treatment of severe, life-threatening diseases, today announced that Wendy Levin, M.D., M.S. will join the Company as Vice President, Clinical Development and that Walter Grubb has joined the Company as Vice President, Business Development. Additionally, the Company announced the promotion of John Ferraro to the newly-created position of Vice President, Clinical Operations.
"We are delighted to welcome Wendy and Walter, two accomplished life sciences industry professionals, to our team, and for John to assume additional leadership responsibilities as head of our clinical operations group," said Christian Weyer, M.D., M.A.S., President and Chief Executive Officer of Fate Therapeutics. "The broad experience and expertise of these leaders will be invaluable as we continue to advance a R&D and business development strategy aimed at building a robust pipeline of first-in-class programmed hematopoietic cellular therapeutics for a range of severe, life-threatening disorders."
Dr. Levin will play a critical role spearheading the Company's clinical development of PROHEMA® in patients with hematologic malignancies and rare genetic disorders. Dr. Levin, a board-certified hematologist / oncologist with extensive experience in clinical oncology research, joins Fate Therapeutics from MEI Pharma, where she was Vice President of Clinical Development and Medical Affairs. Previously, she held positions of increasing responsibilities at Pfizer from 2007 to 2013, where she led a global oncology program targeting the hedgehog pathway from first-in-human studies into later-stage development. Dr. Levin completed her Internal Medicine Residency at the University of Southern California, and went on to complete a Hematology / Oncology Fellowship at the University of Washington / Fred Hutchinson Cancer Research Center in Seattle.
Mr. Grubb's responsibilities will include the assessment, valuation and licensing of business opportunities for the Company. Mr. Grubb was previously Executive Director of Business Development and Commercial Operations at Ambit Biosciences, where he was responsible for multiple partnership transactions, provided commercial leadership for product candidates spanning a range of indications and stages of development, and was instrumental in the company's initial public offering and ultimate acquisition by Daiichi-Sankyo. Prior to Ambit, Mr. Grubb held positions of increasing responsibility at Neurocrine Biosciences from 2000 to 2008 that included business development, market analytics, medical affairs and product marketing.
Mr. Ferraro joined Fate Therapeutics in February 2013, and will be responsible for the Company's clinical operations and data management functions. Prior to joining Fate, Mr. Ferraro was the Senior Director of Clinical Operations at GlobeImmune, Inc., where he established the clinical operations group and led the advancement of multiple targeted immunotherapies through the clinical development process in the United States and in Europe. Mr. Ferraro's professional experience spans 20 years of biopharmaceutical industry experience in global clinical operations and research positions at Pfizer and SmithKline Beecham.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the development of programmed cellular therapeutics for the treatment of severe, life-threatening diseases. The Company's approach utilizes established pharmacologic modalities, such as small molecules, to program the fate and function of cells ex vivo. The Company's lead product candidate, PROHEMA®, is an ex vivo programmed hematopoietic cellular therapeutic, which is currently in clinical development for the treatment of hematologic malignancies and rare genetic disorders in patients undergoing hematopoietic stem cell transplantation (HSCT). The Company is also using its proprietary induced pluripotent stem cell platform to develop ex vivo reprogrammed hematopoietic and myogenic cellular therapeutics. Fate Therapeutics is headquartered in San Diego, CA. For more information, please visitwww.fatetherapeutics.com.
Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the Company's clinical and development plans with respect to PROHEMA and other product candidates. These and any other forward-looking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, the risk of cessation or delay of any clinical development activities for a variety of reasons (including additional information that may be requested or additional obligations that may be imposed by the FDA, any difficulties or delays in patient enrollment in current and planned clinical trials, and any adverse effects or events or other negative results that may be observed in these trials), or the risk that we are unable to conduct or complete preclinical and clinical activities necessary to advance any additional hematopoietic cellular therapeutic product candidates. For a discussion of other risks and uncertainties, and other important factors, any of which could cause our actual results to differ from those contained in the forward-looking statements, see the risks and uncertainties detailed in the Company's periodic filings with the Securities and Exchange Commission, including but not limited to the Company's Form 10-Q for the quarter ended September 30th, 2014, and from time to time the Company's other investor communications. Fate Therapeutics is providing the information in this release as of this date and does not undertake any obligation to update any forward-looking statements contained in this release as a result of new information, future events or otherwise.
CONTACT:
Renee Leck, Stern Investor Relations, Inc.
212.362.1200, renee@sternir.com
Investigator-Sponsored Trial to Evaluate Combination of G100 With Local Radiation Therapy in Patients With Sarcoma
SEATTLE and SOUTH SAN FRANCISCO, Feb. 11, 2015 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ), a clinicalstage immunotherapy company, today announced the start of a Phase 1 clinical trial of G100 in combination with radiation therapy in patients with metastatic sarcoma. G100 is an investigational immuno-oncology agent designed to generate a robust anti-tumor immune response when administered directly to the tumor micro-environment. The investigator-sponsored trial is being conducted at the Fred Hutchinson Cancer Research Center.
"We look forward to investigating the potential of G100 in combination with radiation therapy to induce an immune response in patients with metastatic soft tissue sarcoma, an aggressive disease with limited treatment options," said Seth Pollack, M.D., Fred Hutchinson Cancer Research Center and principal investigator. "G100 is designed to activate antigen presenting cells (APCs) and boost pre-existing anti-tumor cytotoxic T cells or CTLS. In addition, radiation therapy can lead to lysis of tumor cells that release tumor antigens that can be recognized by APCs. The combination of intra-tumoral G100 with local radiation is expected to boost the immune system's response to tumor antigens in the tumor microenvironment and lead to a broad, systemic anti-tumor response."
The Phase 1 open label trial is designed to evaluate the safety, clinical efficacy and immunogenicity of intra-tumoral G100 in combination with palliative radiation therapy in patients with metastatic soft tissue sarcoma. Per protocol, patients will receive treatment with G100 weekly starting prior to the radiation therapy and continuing for eight weeks, and efficacy will be evaluated based on radiographic response in metastatic lesions. "We are pleased that intra-tumoral administration of G100 is now being actively studied in two indications - metastatic soft tissue sarcoma and Merkel cell carcinoma - both alone and in combination with local radiation," said Richard Kenney, M.D., Chief Medical Officer of Immune Design. "The key component of G100, GLA, has been evaluated in more than 1,000 subjects and has demonstrated the ability to stimulate both the innate and adaptive immune system while being well tolerated."
About G100 and the Endogenous Antigen Approach
G100 is an investigational agent that is a product of the company's GLAASTM discovery platform and includes a specific formulation of Glucopyranosyl Lipid A (GLA), a synthetic, Toll-like Receptor-4 (TLR-4) agonist. G100 is part of Immune Design's "Endogenous Antigen" approach to treating cancer, which leverages the activation of dendritic cells in the tumor microenvironment, potentially to create a robust immune response against the tumor's preexisting diverse set of antigens. Preclinical and clinical data have demonstrated the ability of G100 to activate dendritic cells in tumors and to increase antigendependent systemic humoral and cellular Th1 immune responses. A Phase 1 study of G100 in patients with Merkel cell carcinoma is also currently underway and a Phase 1 study of G100 in combination with local radiation in patients with non- Hodgkin lymphoma is planned for the second quarter of this year.
Clinical Development Beyond G100
In addition to the Endogenous Antigen approach, Immune Design has a separate immuno-oncology approach known as "Specific Antigen" that utilizes its ZVexTM platform to deliver specific tumor antigen genes directly to cancer patients' dendritic cells in vivo. The company is pursuing this approach simultaneously through its LV305 and CMB305 clinical programs. The Specific and Endogenous Antigen programs constitute independent approaches that offer the potential to benefit patients with a wide range of both accessible and inaccessible tumors, as well as the opportunity to be combined with other immunooncology approaches such as checkpoint inhibitors and ex vivo engineered T cells.
For Patients
More information about this study, including additional eligibility criteria and contact information, can be found on clinicaltrials.gov.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to generate and/or expand antigen-specific cytotoxic T cells, while enhancing other immune effectors, to fight cancer and other chronic diseases. Immune Design's three on-going immuno-oncology clinical programs are the product of its two synergistic discovery platforms: ZVexTM and GLAASTM, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Cautionary Note Regarding Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend," "believe" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include statements regarding expectations about the ability of G100 with local radiation to boost the immune system and to generate an anti-tumor response and the timing and likelihood of commencing a third clinical trial evaluating G100 in combination with local radiation in non-Hodgkin lymphoma. Factors that may cause actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission (the "SEC"), including the "Risk Factors" section of Immune Design's Quarterly Report on Form 10-Q filed with the SEC on November 13, 2014 and in any subsequent filings with the SEC. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
CONTACT:
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Robert H. Uhl
Westwicke Partners
robert.uhl@westwicke.com
858-356-5932
Amplifiers to Be Developed Complementing Emerging Standard of Care and Delivering Even Greater Benefit to CF Patients
CAMBRIDGE, MA--(Marketwired - February 04, 2015) - Proteostasis Therapeutics, Inc. (PTI), a company developing novel therapeutics to treat diseases of protein folding, trafficking and clearance, today unveiled a new class of agents, CFTR AMPLIFIERS, for the treatment of cystic fibrosis (CF). CFTR amplifiers represent a new drug class able to enhance the effect of known cystic fibrosis transmembrane conductance regulator (CFTR) modulating agents, such as potentiators and correctors. The amplifiers are effective across CFTR mutation classes and form the basis for Proteostasis' strategy to develop a broad acting combination therapy able to serve CF patients with most mutations.
Proteostasis Therapeutics also announced today that it has nominated PTI130 as a clinical development candidate for the treatment of CF. PTI130, an amplifier, was found to have excellent pharmacologic properties amenable for oral dosing. Fourteen-day, non-GLP preclinical toxicology studies testing multiple dose groups of PTI130 in two different animal species demonstrated a favorable safety and tolerability profile for clinical development.
PTI130 is fully owned by Proteostasis Therapeutics and was internally discovered through the Company's proprietary Disease-Relevant Translation, DRT™ platform. DRT™ utilizes functionally pertinent phenotypic assays and disease relevant models to identify highly translatable therapies associated with the modulation of protein homeostasis pathways within the cell. PTI130 was derived from medicinal chemistry optimization of hits from the Company's internal compound library based on its impressive ability to double the efficacy of CFTR modulating agents such as correctors and potentiators, in the HBE (human bronchial epithelial) cell Ussing functional assay. These data provide a basis for the clinical exploration of adding PTI130 on to the emerging standard of care (corrector/potentiator combination) to deliver even greater benefit to CF patients. The discovery of PTI130 was partially funded by research grants from Cystic Fibrosis Foundation Therapeutics Inc., the nonprofit affiliate of the Cystic Fibrosis Foundation.
Proteostasis Therapeutics is expected to file an IND for PTI130 with the US FDA and the molecule will enter the clinic in phase I trials by the end of 2015.
"We are proud of our drug discovery platform that has allowed us to identify molecules with novel mechanisms of action such as CFTR amplifiers," said Meenu Chhabra, President and Chief Executive Officer. "We believe that PTI130 places amplifiers on the vanguard of the rapidly evolving CF treatment paradigm and may prove to be the lynchpin for maximizing benefit for all CF patients regardless of the underlying CFTR mutation class."
About Cystic Fibrosis
Cystic fibrosis is the most common genetic disorder affecting approximately 70,000 people worldwide. Improvement in disease management protocols and approval of new drugs to treat the symptoms have extended the life expectancy for CF patients and is now approaching 40 years of age. However, CF remains an incurable disease that leads to death. Recently, CFTR mutation specific disease modulators such as correctors and potentiators, have shown the ability to restore CFTR function in selected genotypes.
About Proteostasis Therapeutics
Proteostasis Therapeutics is developing disease-modifying therapeutics for diseases of protein folding, trafficking and clearance. By combining the DRT™ platform with state of the art medicinal chemistry tools, PTI generates highly selective drug candidates. The Company's proprietary Disease-Relevant Translation, DRT™ platform utilizes functionally pertinent phenotypic assays and disease relevant models to identify highly translatable therapies associated with the modulation of proteostasis pathways within the cell. PTI has multiple wholly owned programs in cystic fibrosis. PTI collaborates with Biogen Idec to research and develop therapeutic candidates for neurodegenerative disease based on the inhibition of Usp14, which may result in total payments of up to $200M plus royalties. In addition, PTI has a collaboration with Astellas Inc. based on the Unfolded Protein Response (UPR) pathway where PTI is eligible to earn up to $1.2B in development milestones.
For more information visit www.proteostasis.com.
Investor Contact:
Malini Chatterjee
Blueprint Life Science Group
mchatterjee@bplifescience.com
(917) 330-4269
- LTI received funding for preclinical advancement of its Parkinon’s disease therapeutic candidate and development of a biomarker package to guide clinical development.
- LTI appointed Darren Braccia as vice president of business development to lead external collaboration efforts and to aid in the expansion of LTI’s platform by exploring known targets with genetic links between lysosomal storage disorders and neurodegenerative diseases
CAMBRIDGE, Mass. — Feb. 3, 2015 — Lysosomal Therapeutics Inc. (LTI), a company leveraging its expertise in lysosomal biology to develop novel small molecules for use in the treatment of neurodegenerative diseases, announced today it has raised $20 million in Series A financing. This internal round consisted of expanded funding commitments from its existing investors: Atlas Venture, Hatteras Venture Partners, Lilly Ventures, Sanofi-Genzyme BioVentures, Roche Venture Fund, Partners Innovation Fund and several original angel investors, including Orion Equity Partners, LLC, and LTI co-founders Henri Termeer and Bob Carpenter.
The proceeds will fund ongoing compound optimization and preclinical development of a glucocerebrosidase (GCase) lysosomal enzyme activator candidate for the treatment of Parkinson’s disease. In addition, the funding will support a biomarker initiative, for which LTI has previously received grant funding from The Michael J. Fox Foundation for Parkinson’s Research. This biomarker effort will help LTI select patients for future clinical trials and predict drug response. LTI will also initiate new research programs based on the genetic links that exist between other lysosomal storage disorders and neurodegenerative diseases.
“LTI succeeded in meeting its initial, seed-stage goal – to generate a compelling lead-stage molecule within its GCase program,” said Bruce Booth, Ph.D., partner at Atlas Venture, the lead investor of LTI’s Series A financing. “With the syndicate’s participation in this Series A round, we are not only supporting further development of LTI’s breakthrough therapeutic mechanism for the treatment of Parkinson’s disease, but we are also enabling LTI to expand beyond Parkinson’s disease and build out a platform around additional novel targets implicated in underserved orphan and neurodegenerative disorders.”
To help guide LTI’s platform expansion and partnering strategy, the company has hired Darren Braccia as vice president of business development. Braccia brings to LTI’s team more than 16 years of business and corporate development experience: He has worked in progressive business development roles for private and public companies, including Biogen Idec, Magellan Biosciences and most recently, at Vertex Pharmaceuticals as senior director of business development. Braccia is a graduate of the Kellogg Graduate School of Management at Northwestern University and holds a bachelor’s degree from Washington and Lee University.
“Our work to date has affirmed our theory that rare diseases, like Gaucher disease, can be used as model systems for developing therapeutics for common neurodegenerative disorders, such as Parkinson’s disease,” said Kees Been, LTI’s founding president and chief executive officer. “With the continued support of our investors and through the expansion of our team, we will advance our Parkinson’s disease therapeutic candidate to a clinical start and also grow our target-rich biology platform by exploring the known relationships between other lysosomal storage and neurodegenerative disorders.”
About the Implications of Lysosomal Storage Disorders on Neurodegenerative Diseases
Lysosomal storage disorders (LSDs) are a group of approximately 60 known genetically inherited diseases characterized by a deficiency of various vital enzymes. All LSDs consist of neurological components, but Gaucher disease (GD) is the most common LSD, occurring when the gene that encodes the lysosomal enzyme glucocerebrosidase (GCase) is mutated and unable to effectively break down its substrate, glucosylceramide. This results in a build-up of lipids in patients’ cells, causing serious health issues.
Recent genetic research suggests that GCase mutations may also cause a predisposition to Parkinson’s disease (PD). The manifestation of the neurotoxic aggregation of the protein alpha-synuclein, also known as Lewy bodies, is the hallmark symptom of PD. Lysosomal Therapeutics Inc.’s (LTI) initial research shows that restoring lysosomal function in human neurons of GD and PD patients may normalize the otherwise-elevated levels of alpha-synuclein. The company is currently developing small molecules that cross the blood-brain barrier and increase GCase activity to potentially treat the root cause – not only the symptoms – of PD. In addition to its work with GD and PD, LTI is investigating other lysosomal enzyme deficiencies and their respective genetic links to common neurodegenerative diseases.
About Lysosomal Therapeutics Inc.
Lysosomal Therapeutics Inc. (LTI) is dedicated to innovative small-molecule research and development in the field of neurodegeneration, yielding new treatment options for patients with severe neurological diseases. Our strategy leverages the clinically validated link between lysosome-based genetic disorders and neurodegenerative diseases to establish a unique and effective molecular platform for novel drug discovery. LTI’s lead program targets Gaucher-related neurodegeneration, Parkinson’s disease and other synucleinopathies. www.lysosomaltx.com
Company Contact Media Contact
Kees Been Lynnea Olivarez
CEO MacDougall Biomedical Communications
(617) 913-0166 (781) 235-3060
Kees.Been@LysosomalTx.com LOlivarez@MacBioCom.com
# # #
Primary Endpoint Read-outs from Adult PUMA and Pediatric PROMPT Studies of PROHEMA in Hematologic Malignancies Expected in mid-2015
Topline Data from Pediatric PROVIDE Study of PROHEMA in Rare Inherited Metabolic Disorders Expected in 2015
IND Filing for New Programmed Mobilized Peripheral Blood Candidate Planned in 2015
SAN DIEGO, Jan. 12, 2015 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE), a biopharmaceutical company engaged in the development of programmed cellular therapeutics for the treatment of severe, life-threatening diseases, today announced key pipeline objectives for 2015.
"Building upon our solid progress in 2014, we are poised to reach multiple clinical data read-outs throughout 2015 and establish the disease-transforming potential of PROHEMA® across a wide range of blood cancers and rare genetic disorders," said Christian Weyer, M.D., M.A.S., President and Chief Executive Officer of Fate Therapeutics. "In late 2014, we reported favorable interim clinical data from our ongoing Phase 2 PUMA study and exciting preclinical findings with our newly-discovered small molecule modulator combination, further strengthening our conviction in the therapeutic potential of ex vivo programmed hematopoietic cells. Continuing this momentum, we have begun to advance a programmed mobilized peripheral blood candidate towards a planned 2015 IND filing, and are actively researching novel mechanisms to enhance the immunotherapeutic potential of CD34+ and T cells. We believe we are well-positioned in the coming year to expand our programmed hematopoietic cellular therapeutics pipeline and to leverage our ex vivo programming approach to pursue significant therapeutic opportunities."
Key 2015 Pipeline Objectives
- Report Full Data on the Primary Efficacy Endpoint in mid-2015 from the Phase 2 PUMA Study of PROHEMA. Fate Therapeutics is currently enrolling adult subjects in its Phase 2 PUMA study of PROHEMA, an ex vivo programmed hematopoietic cellular therapeutic derived from umbilical cord blood. The PUMA study is a randomized, controlled, openlabel clinical trial designed to enroll 60 adult subjects undergoing double umbilical cord blood transplantation for the treatment of blood cancers. The primary objective of the study is to evaluate neutrophil engraftment, which is essential for the successful reconstitution of a new blood and immune system. The study will also assess other key clinical endpoints that contribute to the overall morbidity and mortality of hematopoietic stem cell transplantation (HSCT) including platelet engraftment, engraftment failure, bacterial infections, viral reactivation, graft-versus-host disease, relapse, non-relapse mortality and disease-free survival. The Company expects to report full data on the primary endpoint in mid-2015.
- Report Topline Data in 2015 from the Phase 1b PROVIDE Study of PROHEMA. Fate Therapeutics is currently initiating the clinical investigation of PROHEMA as a cellular enzyme replacement therapy for pediatric patients with inherited metabolic disorders (IMDs). The Company's Phase 1b PROVIDE study is an open-label clinical trial designed to enroll 12 pediatric subjects undergoing single umbilical cord blood transplantation. Sixteen different types of lysosomal and peroxisomal storage diseases, such as Hurler and Hunter syndromes, Krabbe disease and various other leukodystrophies, qualify for treatment under the study's inclusion criteria. The primary objective of the study is to evaluate neutrophil engraftment. The study will also include a series of neuro-imaging and neuro-cognitive assessments to explore the potential of the programmed hematopoietic cells to provide long-term replacement of an otherwise deficient enzyme to the central nervous system through a process known as cross-correction. In in vivo murine models of allogeneic HSC transplantation, Fate Therapeutics has demonstrated that the use of programmed CD34+ cells, as compared to unmodulated cells, led to a significant increase both in the engraftment of donor HSCs and in the donorderived expression of enzyme in the brain. The Company expects to report topline data from the PROVIDE study in 2015.
- Report Full Data on the Primary Efficacy Endpoint in mid-2015 from the Phase 1b PROMPT Study of PROHEMA. The Company is currently enrolling pediatric subjects in its Phase 1b PROMPT study of PROHEMA. The PROMPT study is an open-label clinical trial designed to enroll 18 pediatric subjects undergoing single umbilical cord blood transplantation for the treatment of blood cancers. The primary objective of the study is to evaluate neutrophil engraftment. The Company expects to report full data on the primary endpoint in mid-2015.
- File an Investigational New Drug Application in 2015 for an Ex Vivo Programmed Mobilized Peripheral Blood Candidate. Preclinical data presented by company scientists at the 56 th Annual Meeting and Exposition of the American Society of Hematology in December 2014 showed that the newly-identified small molecule modulator combination of FT1050 and FT4145 enhances the biological properties and the in vivo therapeutic potential of mobilized peripheral blood. The programming of CD34+ cells with FT1050 and FT4145 resulted in a 60-fold increase in CXCR4 gene expression levels and a statistically significant increase in engraftment as compared to unmodulated cells, and T-cells programmed with FT1050 and FT4145 were found to have a 66% reduction of cell-surface protein expression of ICOS, a key T-cell activation marker, and a statistically significant reduction in proliferation rates relative to unmodulated cells. Fate Therapeutics expects to file an Investigational New Drug application in 2015 to assess the safety and efficacy of programmed mobilized peripheral blood in subjects undergoing allogeneic HSCT for the treatment of hematologic malignancies.
- Advance Preclinical Pipeline of Programmed Cellular Therapeutics. Through internal research efforts, Fate Therapeutics continues to target biological mechanisms that are therapeutically-relevant for modulation and to pursue several attractive opportunities for programmed cellular candidates with disease-transforming potential, including programmed CD34+ cells and programmed T-cells for the regulation of the immune system. In 2015, Fate Therapeutics aims to nominate at least one programmed hematopoietic cellular candidate for further development.
Fate Therapeutics to Present at 7th Annual Biotech Showcase
Dr. Weyer will present today an overview of the Company's programs at the 7th Annual Biotech Showcase conference at 11:30 am PST in San Francisco, California. A live webcast of the presentation can be accessed under "Events & Presentations" in the Investors and Media section of the Company's website at www.fatetherapeutics.com. An archived replay of the webcast will be available on the Company's website for 30 days after the conference.
About PROHEMA®
PROHEMA® is an ex vivo programmed hematopoietic cellular therapeutic derived from umbilical cord blood. PROHEMA is produced by modulating the biological properties of hematopoietic stem cells (HSCs) and T-cells of umbilical cord blood ex vivo using the small molecule FT1050 (16,16 dimethyl prostaglandin E2, or dmPGE2). The proprietary modulation process induces rapid activation of genes involved in the homing, proliferation and survival of HSCs and the cell cycle, immune tolerance and anti-viral properties of T-cells. PROHEMA is currently being developed as a donor-derived source of hematopoietic cells for use in allogeneic hematopoietic stem cell transplantation (HSCT) to treat severe, life-threatening hematologic malignancies and inherited metabolic disorders. In 2010, the FDA granted PROHEMA orphan designation for the enhancement of HSC engraftment in patients undergoing umbilical cord blood transplantation.
About Hematopoietic Stem Cell Transplantation
Allogeneic hematopoietic stem cell transplantation (HSCT) is an established procedure performed with curative intent in patients with a wide range of hematologic malignancies and inherited immune and blood disorders. The procedure involves transferring donor-derived hematopoietic cells, including stem cells and T-cells, to a patient following the administration of chemotherapy and/or radiation therapy. Donor-derived hematopoietic stem cells (HSCs) and T-cells each play an essential role in allogeneic HSCT - HSCs reconstitute and replace the patient's blood and immune system, and T-cells act as an immunotherapeutic to eliminate residual malignancy. More than 20,000 allogeneic HSCTs are performed annually worldwide in an effort to achieve long-term disease-free remission and/or functional cures.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the development of programmed cellular therapeutics for the treatment of severe, life-threatening diseases. The Company's approach utilizes established pharmacologic modalities, such as small molecules, to program the fate and function of cells ex vivo. The Company's lead product candidate, PROHEMA®, is an ex vivo programmed hematopoietic cellular therapeutic, which is currently in clinical development for the treatment of hematologic malignancies and rare genetic disorders in patients undergoing hematopoietic stem cell transplantation (HSCT). The Company is also using its proprietary induced pluripotent stem cell platform to develop ex vivo reprogrammed hematopoietic and myogenic cellular therapeutics. Fate Therapeutics is headquartered in San Diego, CA. For more information, please visit www.fatetherapeutics.com.
Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the therapeutic potential of PROHEMA® and programmed mobilized peripheral blood, the Company's plans with respect to PROHEMA and other product candidates, anticipated clinical and development milestones (including the timing and results of ongoing and planned clinical trials, and the availability of clinical data and results), and the plans of the Company to undertake certain research and development activities, including the evaluation of the ex vivo programming of CD34+ cells and T-cells. These and any other forward-looking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, the risks that the results of PROHEMA observed in prior preclinical and clinical development may not be replicated in current or subsequent clinical trials of PROHEMA, the risk of cessation or delay of any clinical development activities for a variety of reasons (including additional information that may be requested or additional obligations that may be imposed by the FDA, any difficulties or delays in patient enrollment in current and planned clinical trials, and any adverse effects or events or other negative results that may be observed in these trials), or the risk that we are unable to conduct or complete preclinical and clinical activities necessary to advance any additional hematopoietic cellular therapeutic product candidates, including any candidates derived from mobilized peripheral blood, CD34+ cells or T-cells. For a discussion of other risks and uncertainties, and other important factors, any of which could cause our actual results to differ from those contained in the forward-looking statements, see the risks and uncertainties detailed in the Company's periodic filings with the Securities and Exchange Commission, including but not limited to the Company's Form 10-Q for the quarter ended September 30th, 2014, and from time to time the Company's other investor communications. Fate Therapeutics is providing the information in this release as of this date and does not undertake any obligation to update any forward-looking statements contained in this release as a result of new information, future events or otherwise.
CONTACT:
Renee Leck, Stern Investor Relations, Inc.
212.362.1200, renee@sternir.com

NOVATO, Calif., Jan. 12, 2015 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (Nasdaq:RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced its intent to file a Marketing Authorization Application (MAA) seeking conditional approval from the European Medicines Agency (EMA) for the use of six grams per day of sialic acid extended-release (SA-ER; UX001) tablets in the treatment of hereditary inclusion body myopathy (HIBM; also known as GNE myopathy). SA-ER is designed to replace the deficient sialic acid substrate in patients with HIBM, a rare, progressive muscle-wasting disease. Based on Scientific Advice recently received from the EMA's Committee for Medicinal Products for Human Use (CHMP), the company intends to file an MAA in the second half of 2015 for stabilization of upper extremity muscle strength.
"HIBM is a devastating disease that can lead to severe progressive and irreversible muscle damage," said Sunil Agarwal, M.D., Chief Medical Officer of Ultragenyx. "Based on the Phase 2 data demonstrating slowing of disease progression, we are pursuing conditional approval of SA-ER in the European Union in order to accelerate access to this therapy for patients who otherwise have no approved treatment options."
The EMA may grant conditional marketing authorization when the potential treatment addresses a severely debilitating disease with an unmet medical need, has a positive benefit to risk profile, and the benefits to public health of its immediate availability outweigh the risks inherent in the fact that additional data are still required. Ongoing or new studies must be completed with the objective of confirming that the benefit to risk balance is positive. The approval is renewed on an annual basis until all obligations have been fulfilled, at which point a full approval may be granted.
In order to satisfy the EMA's requirement for additional controlled data, Ultragenyx plans to initiate a global, randomized, double-blind, placebo-controlled Phase 3 study of six grams per day of SA-ER in patients with HIBM in mid-2015. A composite of upper extremity muscle strength (UEC) will be the primary endpoint. Key secondary endpoints include GNE myopathyfunctional activity scale (GNEM-FAS) (including patient-reported outcome scores of mobility and upper extremity function), and several measures of lower extremity muscle strength including the lower extremity muscle strength composite (LEC). The US Food and Drug Administration (FDA) has accepted this same Phase 3 study design as pivotal, including the primary endpoint of UEC.
About Hereditary Inclusion Body Myopathy
Hereditary inclusion body myopathy (HIBM) is also known as GNE myopathy. HIBM is a rare, severe, progressive, genetic neuromuscular disease caused by a defect in the biosynthetic pathway for sialic acid, with onset in the late teens or twenties. The body's failure to produce enough sialic acid causes muscles to slowly waste away and can lead to very severe disability, with patients typically becoming wheelchair bound and losing most major muscle function within ten to 20 years from onset. There are approximately 2,000 HIBM patients in the developed world, and there is currently no approved therapy.
About SA-ER Treatment in Hereditary Inclusion Body Myopathy
A Phase 2 randomized, double-blind, placebo-controlled study with SA-ER has been completed. The data showed a statistically significant difference in the upper extremity composite of muscle strength at 48 weeks with a higher dose group compared to a lower dose group. SA-ER appeared to be generally safe and well-tolerated with no serious adverse events observed to date. Over an approximate two-year treatment period in the Phase 2 study and long-term extension, SA-ER appeared to slow the progression of upper extremity disease when compared to the 24-week placebo group extrapolated out to two years.
About Ultragenyx
Ultragenyx is a clinical-stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with a focus on serious, debilitating genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
Forward-Looking Statements
Except for the historical information contained herein, the matters set forth in this press release, including statements regarding the intent to file an MAA and the anticipated timing of such filing, as well as plans for a potential pivotal study and the timing of same, are forward-looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance, or achievements to differ significantly from those expressed or implied by the forward-looking statements. Such risks and uncertainties include, among others, the uncertainties inherent in the clinical drug development process, including the regulatory approval process, the timing of our regulatory filings, and other matters that could affect the availability or commercial potential of our drug candidate. Ultragenyx undertakes no obligation to update or revise any forward-looking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 10, 2014, and its subsequent periodic reports filed with the Securities and Exchange Commission.
CONTACT:
Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
NOVATO, Calif., Jan. 7, 2015 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (Nasdaq:RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced a license agreement with Inserm Transfert SA and Institut du Cerveau et de la Moelle Epiniere (ICM) for intellectual property related to the treatment of Huntington's disease with triheptanoin (UX007).
"Huntington's is a severe and lethal rare genetic disease," commented Emil D. Kakkis, Ph.D., M.D., Chief Executive Officer and President of Ultragenyx. "Energy deficiency is believed to play a role in the pathophysiology of Huntington's disease, and we are encouraged by data from a small pilot study suggesting improvement in brain energy metabolism after treatment with triheptanoin."
"This pilot study, as well as previous metabolic studies that we performed, indicate that brain energy deficit is a realistic therapeutic target in Huntington's disease," said Fanny Mochel, M.D., Ph.D., principal investigator of the study at ICM.
Ultragenyx is supporting a second investigator-sponsored clinical study being planned by ICM to evaluate the safety and efficacy of triheptanoin in patients with Huntington's disease.
About Huntington's Disease
Huntington's disease is an autosomal dominant neurodegenerative disease characterized by movement disorders (chorea), behavioral and psychiatric disturbances, dementia, and death. The clinical features of the disease usually emerge between 30 and 50 years of age. The only approved therapy is indicated for the treatment of chorea associated with the disease. There are an estimated 30,000 patients with Huntington's disease in the U.S.
A pilot study was completed in ten patients with early stage Huntington's disease in which triheptanoin appeared to impact brain energy metabolism and the Unified Huntington's Disease Rating Scale motor score. Results from the study were published in the January 7, 2015 online issue of Neurology, the medical journal of the American Academy of Neurology.
About Triheptanoin
Triheptanoin, also known as UX007, is a purified, pharmaceutical-grade, specially designed synthetic triglyceride compound created via a multi-step chemical process. Ultragenyx is currently evaluating triheptanoin in two clinical programs.
The first program is studying the genetic seizure disorder Glut1 deficiency syndrome (Glut1 DS). This disease is caused by a genetic defect in the transport of glucose into the brain and affects an estimated 3,000 to 7,000 patients in the U.S. Glut1 DS is characterized by seizures, developmental delay, and movement disorder. Triheptanoin is metabolized to heptanoate, which can diffuse across the blood-brain barrier and be converted into glucose. Heptanoate can also be further metabolized in the liver to four- and five-carbon ketone bodies that also cross the blood-brain-barrier. Both are intended to provide patients with an additional energy source to the brain. Heptanoate and five-carbon ketone bodies can also regenerate new glucose in the brain, which is deficient in these patients. Ultragenyx is conducting a Phase 2 study in the U.S. and Europe to evaluate the potential of triheptanoin to treat Glut1 DS patients who are not on or who have failed the ketogenic diet and continue to have seizures.
The second program is studying long-chain fatty acid oxidation disorders (LC-FAOD), a set of rare metabolic diseases caused by the inability to convert fat into energy, leading to low blood sugar, muscle rupture, and heart and liver disease. Triheptanoin is intended to provide patients with medium-length, odd-chain fatty acids that can be metabolized to increase intermediate substrates in the Krebs cycle, a key energy-generating process. Unlike typical even-chain fatty acids, triheptanoin can be converted to new glucose through the Krebs cycle, potentially providing an important added therapeutic effect, particularly when glucose levels are too low. Ultragenyx is conducting a Phase 2 study to evaluate the potential of triheptanoin to treat LCFAOD.
About Ultragenyx
Ultragenyx is a clinical-stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with a focus on serious, debilitating genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies. The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
Forward-Looking Statements
Except for the historical information contained herein, the matters set forth in this press release, including statements regarding a planned investigator-sponsored clinical study, are forward-looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance, or achievements to differ significantly from those expressed or implied by the forward-looking statements. Such risks and uncertainties include, among others, the uncertainties related to timing and results of the planned investigator-sponsored clinical study, whether or not we ever pursue our own clinical study and whether or not we ever seek to obtain approval for the use of triheptanoin to treat Huntington's disease, as well as uncertainties inherent in the clinical drug development process, including the regulatory approval process, the timing of our regulatory filings, and other matters that could affect the availability or commercial potential of our drug candidates. Ultragenyx undertakes no obligation to update or revise any forward-looking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's Quarterly Report on Form 10- Q filed with the Securities and Exchange Commission on November 10, 2014, and its subsequent periodic reports filed with the Securities and Exchange Commission.
CONTACT:
Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
Key Metrics of Neutrophil Engraftment Improved in PROHEMA Arm
Independent Data Monitoring Committee Supports Continuation of Study Enrollment
Full Results on Primary Efficacy Endpoint Expected in mid-2015
SAN DIEGO, Dec. 18, 2014 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE), a biopharmaceutical company engaged in the discovery and development of adult stem cell modulators to treat orphan diseases, today reported initial data from the first 12 subjects administered PROHEMA® in the Company's ongoing Phase 2 PUMA study and announced that the study's independent Data Monitoring Committee (iDMC) supported continuation of the clinical trial following a second planned interim safety review. These early data showed that subjects administered PROHEMA, the Company's lead ex vivo programmed hematopoietic cellular therapeutic derived from umbilical cord blood, had an improved median time of neutrophil engraftment and an increased incidence of early neutrophil engraftment.
"Our evaluation of these initial data from 12 subjects shows that small molecule-based programming of hematopoietic stem cells and T cells ex vivo can positively affect the biological activity of these cells in vivo. Given that many of the complications contributing to the 25-30% early mortality following allogeneic hematopoietic stem cell transplantation are tied to the function of donor-derived HSCs and T cells, we believe our ex vivo programming paradigm can meaningfully improve outcomes of this potentially curative procedure," said Christian Weyer, M.D., M.A.S., President and Chief Executive Officer of Fate Therapeutics. "In 2015, we expect to report on multiple clinical milestones in our PROHEMA development program, including full results on the primary and secondary endpoints of this PUMA study, as well as clinical data read-outs from our PROMPT and PROVIDE studies in pediatric patients with hematologic malignancies and inherited metabolic disorders, respectively."
PROHEMA is derived by programming the biological properties of allogeneic hematopoietic cells ex vivo using a small molecule pharmacologic modulator, optimizing their in vivo therapeutic potential. Of the first 12 subjects administered PROHEMA in the Phase 2 PUMA study, 10 subjects received myeloablative conditioning (MAC) and two subjects received reduced-intensity conditioning (RIC). Eight of 10 PROHEMA subjects receiving MAC achieved neutrophil engraftment, with a median time of engraftment of 20 days. One of two PROHEMA subjects receiving RIC achieved neutrophil engraftment, which was reached on Day 14. Historical median times of neutrophil engraftment are approximately 26 days for patients receiving MAC and 21 days for patients receiving RIC based on multi-center reports published in the literature of adult patients undergoing double umbilical cord blood transplantation in the United States. Six of the nine engrafting subjects administered PROHEMA in the PUMA study achieved neutrophil engraftment prior to these historical median times.
The engraftment of donor-derived hematopoietic stem cells (HSCs) in patients undergoing hematopoietic stem cell transplantation (HSCT) is essential for the successful reconstitution of a new blood and immune system. Complications from delayed or failed neutrophil engraftment following allogeneic HSCT are a leading contributor to non-relapse mortality, and this risk increases by 2.5-fold in patients with delayed neutrophil recovery. The PUMA study is designed to evaluate the effect of PROHEMA on hematopoietic reconstitution and immune-mediated outcomes across multiple clinical endpoints that contribute significantly to the overall morbidity and mortality of allogeneic HSCT. These include time to and incidence of neutrophil and platelet engraftment, engraftment failure, bacterial infections, viral reactivation, graft versus host disease, relapse of underlying disease and overall and disease-free survival.
On December 16, 2014, the PUMA study's iDMC conducted its second of two scheduled interim safety reviews of PROHEMA. A total of 20 subjects, including 12 subjects that received PROHEMA plus an unmanipulated cord blood unit and eight control subjects that received two unmanipulated cord blood units, were included in the interim review, which assessed safety, time to engraftment, rates of graft failure, early mortality, infection and graft versus host disease. Two early deaths prior to engraftment, which were both attributed to the toxicity of the conditioning regimen received by the subjects, were reported in the PROHEMA arm, and one subject administered PROHEMA failed to achieve neutrophil engraftment. Based on its consideration of the data available on the first 20 subjects as well as historical outcomes reported from multi-center clinical experiences, the iDMC determined that PROHEMA had met established safety criteria and supported continuation of the PUMA study. The Company believes that data from its Phase 2 control subjects are consistent with expected outcomes of patients undergoing double umbilical cord blood transplantation published in the literature, including historical median times of neutrophil engraftment.
"There are an estimated 15,000 patients in need of a donor-derived transplant each year, and up to 40 percent of these patients do not have access to a matched donor source. Given its rapid accessibility and less stringent HLA matching requirements, as well as the significantly lower rates of relapse reported following its clinical use, umbilical cord blood is an advantageous source of hematopoietic cells for allogeneic transplantation," said Pratik Multani, M.D., Chief Medical Officer of Fate Therapeutics. "Initial data on neutrophil engraftment from the PUMA study provide encouragement that PROHEMA may address delayed hematopoietic reconstitution, which is the most significant barrier to umbilical cord blood's broad clinical use as a potentially curative treatment for patients with hematologic malignancies and severe inherited blood and immune disorders."
In addition to its conduct of the PUMA study, Fate Therapeutics is currently investigating the therapeutic potential of PROHEMA in a Phase 1b clinical trial in pediatric patients with hematologic malignancies (PROMPT), and plans to initiate a Phase 1b clinical trial of PROHEMA in pediatric patients with inherited metabolic disorders (PROVIDE).
About PROHEMA®
PROHEMA® is an ex vivo programmed hematopoietic cellular therapeutic derived from umbilical cord blood. PROHEMA is produced through a proprietary, two-hour programming process, where the biological properties of hematopoietic stem cells (HSCs) and T cells of umbilical cord blood are modulated ex vivo using a small molecule pharmacologic modulator to optimize these cells' therapeutic potential in vivo. The programming process, which uses the small molecule pharmacologic modulator FT1050 (16,16 dimethyl prostaglandin E2, or dmPGE2), induces rapid activation of genes involved in the homing, proliferation and survival of HSCs and the cell cycle, immune tolerance and anti-viral properties of T cells. PROHEMA is currently being developed as a donor source of hematopoietic cells for use in allogeneic HSCT for the treatment of a wide range of hematologic malignancies in adult and pediatric patients and for the treatment of a wide range of inherited metabolic disorders in pediatric patients. In 2010, the FDA granted PROHEMA orphan designation for the enhancement of HSC engraftment in patients undergoing umbilical cord blood transplantation.
About the Phase 2 PUMA Study
The PUMA (PROHEMA® in UMbilical cord blood transplant in Adults) study is a randomized, controlled, open-label Phase 2 clinical trial of PROHEMA in adult subjects undergoing double umbilical cord blood transplantation for the treatment of hematologic malignancies including acute lymphoblastic leukemia (ALL), acute myelogenous leukemia (AML) and non-Hodgkin lymphoma (NHL). Multiple clinical endpoints that contribute significantly to the overall morbidity and mortality of allogeneic HSCT are being investigated in the PUMA study to inform and support potential registrational strategies. These clinical endpoints include key measures of the hematopoietic reconstitution and immunotherapeutic potential of PROHEMA including time to and incidence of neutrophil and platelet engraftment, engraftment failure, bacterial infections, viral reactivation, graft versus host disease, relapse of underlying disease and overall and disease-free survival.
The Phase 2 PUMA study is designed to enroll 60 subjects. Eligible subjects are randomized, at a ratio of 2:1, with approximately 40 subjects intended to receive PROHEMA plus an unmanipulated cord blood unit, and approximately 20 subjects intended to receive two unmanipulated cord blood units. Based upon physician choice, subjects are treated with one of two conditioning regimens, an intense myeloablative regimen or a reduced-intensity regimen, to destroy malignant cells and to prevent rejection of the donor hematopoietic cells. Randomization will be stratified by conditioning regimen. An independent Data Monitoring Committee is providing safety oversight during the conduct of the PUMA study.
The primary endpoint of the 60-subject Phase 2 PUMA study is based on a categorical analysis of neutrophil engraftment, and the clinical trial is powered to show with statistical significance that 70% of subjects with neutrophil engraftment in the PROHEMA treatment arm engraft prior to a pre-specified control day of neutrophil engraftment. The pre-specified control day of neutrophil engraftment, which is dependent on the conditioning regimen received by the subject, has been established as 26 days for subjects receiving myeloablative conditioning and 21 days for subjects receiving reduced-intensity conditioning. These pre-specified values are based on multi-center reports published in the literature of historical median times to neutrophil engraftment in adult patients undergoing double umbilical cord blood transplantation in the United States (Brunstein, Blood. 2010;116(22): 4693-4699; Cutler, Bone Marrow Transplantation. 2011;46(5): 659-67). The Phase 2 PUMA study utilizes the concurrent control arm to validate the pre-specified values of neutrophil engraftment and to provide context for interpretation of other clinical outcomes. Full data on the primary efficacy endpoint from the Phase 2 PUMA study are expected in mid-2015.
About Hematopoietic Stem Cell Transplantation
Allogeneic hematopoietic stem cell transplantation (HSCT) is an established procedure performed with curative intent in patients with a wide range of hematologic malignancies and life-threatening inherited immune and blood disorders. More than 20,000 allogeneic HSCTs are performed annually worldwide to achieve long-term disease-free remission and/or functional cures. The majority of HSCTs are performed in adults for the treatment of hematologic malignancies and in children for the treatment of inherited disorders. Disease-free survival rates of approximately 40-50% at two- and five-years following HSCT have been reported in multi-center clinical experiences for the treatment of hematologic malignancies.
The procedure involves transferring donor-derived hematopoietic cells, including stem cells and T cells, from mobilized peripheral blood, bone marrow or umbilical cord blood to a patient following the administration of chemotherapy and/or radiation therapy. Donor-derived hematopoietic stem cells (HSCs) and T cells each play an essential role in allogeneic HSCT - donor HSCs traffic to and engraft in the bone marrow, replacing the patient's blood and immune system, and T cells traffic to and engage cancer cells, acting as an immunotherapeutic to eliminate residual malignancy. Delay or failure of HSC engraftment leaves a patient severely immuno-compromised and exposes the patient to significant risk of early mortality. Additionally, while the donor T cells impart a critical immunotherapeutic effect, the reactivity of donor T cells can result in a serious complication known as graft versus host disease, where donor T cells recognize antigens on patient's cells as foreign and attack the patient's cells. While allogeneic HSCT holds curative potential, the majority of relapse and non-relapse mortality occurs within the initial months of the procedure, where the rate of relapse and non-relapse mortality is approximately 25-30% at Day 100 following HSCT.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the discovery and development of pharmacologic modulators of adult stem cells to treat severe, life-threatening diseases. The Company's approach utilizes established pharmacologic modalities, such as small molecules, and targets well-characterized biological mechanisms to program the fate and enhance the therapeutic potential of adult stem cells. The Company's lead product candidate, PROHEMA®, is an ex vivo programmed hematopoietic cellular therapeutic, which is currently in clinical development for patients undergoing HSCT. The Company is also applying its reprogramming modulators to develop human induced pluripotent stem cell-derived cellular therapeutics, and evaluating the in vivo programming of muscle satellite stem cells using its Wnt7a-based protein analogs for muscle regeneration. Fate Therapeutics is headquartered in San Diego, CA. For more information, please visit www.fatetherapeutics.com.
Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the potential safety and efficacy of PROHEMA® (including statements relating to observations of improved median times to engraftment and increased incidence of early engraftment in subjects treated with PROHEMA in the PUMA study), the Company's clinical development plans for PROHEMA and the availability of clinical data and results. These and any other forward-looking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. In particular, the PUMA study is currently ongoing and in the earlier stages of patient enrollment and evaluation, there is limited data concerning treatment with PROHEMA in the PUMA study, and the announced data are preliminary in nature. Additional risks and uncertainties include, but are not limited to, the risks that the results of PROHEMA observed in prior preclinical and clinical development, and in these preliminary data in the PUMA study, may not be replicated in ongoing or future clinical trials of PROHEMA, the risk that any conclusions drawn from preliminary data on a subset of patients may not be supported based on subsequent data, and the risk of cessation or delay of any clinical development activities for a variety of reasons (including additional information that may be requested or additional obligations that may be imposed by the FDA, any difficulties or delays in patient enrollment in current and planned clinical trials, and any adverse events or other negative results that may be observed in these trials). For a discussion of other risks and uncertainties, and other important factors, any of which could cause our actual results to differ from those contained in the forward-looking statements, see the risks and uncertainties detailed in the Company's periodic filings with the Securities and Exchange Commission, including but not limited to the Company's Form 10-Q for the quarter ended September 30th, 2014, and from time to time the Company's other investor communications. Fate Therapeutics is providing the information in this release as of this date and does not undertake any obligation to update any forward-looking statements contained in this release as a result of new information, future events or otherwise.
CONTACT:
Renee Leck, Stern Investor Relations, Inc.
212.362.1200, renee@sternir.com
Agreement Reached With Both FDA and EMA on Pivotal Trial Design
NOVATO, Calif., Dec. 15, 2014 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (Nasdaq:RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced the dosing of the first patient in the pivotal Phase 3 study of recombinant human beta-glucuronidase (rhGUS, UX003), an investigational therapy for the treatment of Mucopolysaccharidosis 7 (MPS 7, Sly syndrome).
"We are pleased to have reached alignment with both the FDA and EMA on our single pivotal study design for this devastating disease," commented Sunil Agarwal, M.D., Chief Medical Officer of Ultragenyx. "This is an important step forward for these patients who have no approved treatment options. It is also an important milestone for Ultragenyx as it is the start of our first Phase 3 program."
The Phase 3 global, randomized, placebo-controlled, blind-start clinical study will assess the efficacy and safety of rhGUS in 12 patients between 5 and 35 years of age. Patients will be randomized to one of four groups. One cohort begins rhGUS therapy immediately, while the other three start on placebo and cross over to rhGUS at different predefined time points in a blinded manner. This novel trial design generates treatment data from all 12 patients, improving the statistical power of the study relative to a traditional parallel-group design. Based on data from the Phase 1/2 study, patients will be dosed with 4 mg/kg of rhGUS every other week for up to a total of 48 weeks, and all groups will receive a minimum of 24 weeks of treatment with rhGUS.
The primary objective of the study is to determine the efficacy of rhGUS as determined by the reduction in urinary GAG excretion after 24 weeks of treatment. The Phase 3 study will also evaluate the safety and tolerability of rhGUS, pulmonary function, walking, stair climb, shoulder flexion, fine and gross motor function, hepatosplenomegaly, cardiac size and function, visual acuity, patient and caregiver assessment of most significant clinical problems, global impressions of change, a multidomain responder index, and other endpoints.
Agreement with the U.S. Food and Drug Administration (FDA) was reached that their evaluation of the pivotal Phase 3 study will be based on the totality of the data on a patient-by-patient basis. FDA advised against the declaration of a primary clinical endpoint in order to allow for more flexibility in the overall efficacy evaluation, appreciating the heterogeneous and rare nature of this disease.
The European Medicines Agency (EMA) has already agreed that approval under exceptional circumstances could be possible based upon a single positive Phase 3 study using urinary GAG excretion as the primary endpoint with a trend toward improvement in the most important clinical endpoints (six-minute walk test, forced expiratory volume, spleen/liver volume).
Data from the Phase 3 study is expected in the first half of 2016. Longer-term 36-week data from the Phase 1/2 study is expected to be presented at the Lysosomal Disease Network's 11th Annual World Symposium in February 2015. Two additional patients continue to be treated under emergency Investigational New Drug (eIND) applications.
About MPS 7
Mucopolysaccharidosis type 7 (MPS 7, Sly syndrome), originally described in 1973 by William Sly, M.D., is a rare genetic, metabolic disorder and is one of 11 different MPS disorders. MPS 7 is caused by the deficiency of beta-glucuronidase, an enzyme required for the breakdown of the glycosaminoglycans (GAGs) dermatan sulfate and heparan sulfate. These complex GAG carbohydrates are a critical component of many tissues. The inability to properly break down GAGs leads to a progressive accumulation in many tissues and results in a multi-system disease.
While its clinical manifestations are similar to MPS 1 and MPS 2, MPS 7 is one of the rarest among the MPS disorders. MPS 7 has a wide spectrum of clinical manifestations and can present as early as at birth. There are no approved therapies for MPS 7 today. The use of enzyme replacement therapy as a potential treatment is based on 20 years of research work in murine models of the disease. Enzyme replacement as a strategy is well established in the MPS field as there are currently four approved enzyme replacement therapies for other MPS disorders: MPS 1 (Aldurazyme®, laronidase), MPS 2 (Elaprase®, idursulfase), MPS 4A (Vimizim™, elosulfase alfa), and MPS 6 (Naglazym®e, galsulfase).
About Ultragenyx
Ultragenyx is a clinical-stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with a focus on serious, debilitating metabolic genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
Forward-Looking Statements
Except for the historical information contained herein, the matters set forth in this press release, including statements regarding expectations regarding the timing of release of data, are forward-looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance, or achievements to differ significantly from those expressed or implied by the forward-looking statements. Such risks and uncertainties include, among others, uncertainties inherent in the clinical drug development process, including the regulatory approval process, the timing of our regulatory filings, and other matters that could affect the availability or commercial potential of our drug candidates. Ultragenyx undertakes no obligation to update or revise any forward-looking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on November 10, 2014, and its subsequent periodic reports filed with the Securities and Exchange Commission.
CONTACT:
Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
CAMBRIDGE, MA--(Marketwired - December 03, 2014) - Proteostasis Therapeutics, Inc., a company developing novel therapeutics that regulate protein homeostasis to improve outcomes for patients with orphan and neurodegenerative diseases, today announced the appointment of Dr. Po-Shun Lee, MD, as Vice President of Clinical Development. Dr. Lee is a pulmonary and critical care physician with extensive experience in biopharmaceutical research and development in both the industry and academia. His initial focus will be on leading the strategic selection process of which of the Company's proprietary PTI CFTR compounds should proceed to clinical development, as part of its leading program in cystic fibrosis.
"Dr. Lee brings to Proteostasis the clinical and managerial drug development expertise that is critical to transitioning our lead program for cystic fibrosis from its current early development to market," commented Meenu Chhabra, Chief Executive Officer of Proteostasis. "As a self-described physician-scientist, Dr. Lee's combined experience will shepherd the Proteostasis pipeline from our proprietary screening platform, through proof of concept, to market, and, most importantly, to the patients suffering from the orphan and neurological diseases we wish to treat."
Prior to joining Proteostasis, Dr. Lee was the Translational Medicine Expert that led the cystic fibrosis and asthma programs from early development to proof of concept at the Novartis Institute for Biomedical Research. Before joining Novartis, Dr. Lee was the Associate Medical Director at Vertex where he supported Kalydeco registration and led a CFTR corrector program to positive proof of concept. Previously, Dr. Lee was a physician-scientist at Brigham Women's Hospital at Harvard Medical School where he was the scientific founder of two biotech companies, Critical Biologics Corporation and BioAegis Therapeutics. He served as a principal investigator in the Pulmonary and Translational Medicine Units, medical director of the Pulmonary Function Lab, an attending physician in the Medical Intensive Care Unit, and Instructor of Medicine at Harvard Medical School. Dr. Lee's academic research interests included endogenous inflammation control, aberrant cellular proliferation in rare lung diseases and gene therapy for cystic fibrosis. He received his medical degree from the University of Pennsylvania School of Medicine and his undergraduate degree from Johns Hopkins University.
"I am honored to join the Proteostasis team at this pivotal moment in their cystic fibrosis program," said Dr. Lee. "Throughout my medical career I have had an interest in and have worked to find different therapies to treat cystic fibrosis. I look forward to helping lead the selection process and guide the design and conduct of future studies that will evaluate the selected drug candidates Proteostasis has so effectively developed with their platform technology."
About Proteostasis Therapeutics Proteostasis
Therapeutics is developing disease-modifying therapeutics for cystic fibrosis, genetic diseases and neurodegenerative diseases. The Company's technology combines both phenotypic and target based drug discovery to develop therapeutics that modulate protein homeostasis pathways and correct for imbalances in protein folding, trafficking, and clearance. For more information, please visit www.proteostasis.com.
Investor Contact:
Malini Chatterjee, PhD
Blueprint Life Science Group
mchatterjee@bplifescience.com
(917) 330-4269
CAMBRIDGE, MA--(Marketwired - November 06, 2014) - Proteostasis Therapeutics, Inc., a company developing novel therapeutics that regulate protein homeostasis to improve outcomes for patients with orphan and neurodegenerative diseases, today announced the publication of its pioneering research study in the field of aging.
This study entitled "A Chaperome Sub-Network Safeguards Proteostasis in Aging and Neurodegenerative Disease" was published in the Nov 6 issue of Cell Reports.
Aging is the most significant and universal risk factor for developing neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS) and Alzheimer's, Parkinson's and Huntington's diseases. This risk increases disproportionately with age, but no one really knows why.
Now a team of scientists from Proteostasis Therapeutics, Inc., Northwestern University and Harvard University has uncovered some clues. The researchers are the first to find that the quality of protective genes called molecular chaperones declines dramatically in the brains of older humans, both healthy and not, and that the decline is further accelerated in humans with neurodegenerative disease.
Chaperones are a class of proteins encoded by a special set of highly conserved genes that watch over cells, keeping them, and the entire organism, healthy by preventing protein damage. If this critical system declines it leads to misfolded and damaged proteins, eventually leading to tissue damage and death. In keeping the chaperones healthy, we may be able to keep a person healthier, even in old age.
The researchers specifically found a decline in 100 genes, corresponding to approximately one-third of all human molecular chaperone genes. Then, with additional studies, they winnowed that number down to 28 human genes specifically involved in age-associated neurodegeneration. These critical genes provide a basis for a biomarker, an early indicator of disease and a target for new therapeutics.
To zero in on the subnetwork of 28 key genes, the scientists combined genomic analysis of human brain tissue, from both healthy individuals and those with neurodegenerative diseases (Alzheimer's, Parkinson's and Huntington's), with functional studies of C. elegans, a transparent roundworm. (The worm is a popular research tool in neurobiology and has provided many insights into human disease.)
Meenu Chhabra, President and CEO of Proteostasis Therapeutics commented that "this pioneering work is particularly notable because it has been able to distill out only 28 key genes from the vast human genome of 25,000 genes and elegantly demonstrate their connection with the aging process. Using our expertise in protein biology we will now be able to delve into the molecular basis for the decline in the encoded chaperones proteins, with the long term goal of developing chaperone targeted therapies to prevent their age associated decline. Preventing or reversing age related neurodegenerative processes is one of the highest unmet medical needs and this research brings us one step closer to the solution and we thank all the scientists involved in this study for their dedication and groundbreaking work."
The primary co-authors of this study are Drs. Marc Brehme and Cindy Voisine. The senior and corresponding authors are Dr. Marc Vidal from Harvard medical School, Dr. Hui Ge, formerly at Proteostasis Therapeutics, and Dr. Richard Morimoto from Northwestern University, who is also a co-founder of and scientific advisory board member at Proteostasis Therapeutics. No funds from the company were used in Dr. Morimoto's laboratory at Northwestern University.
About Proteostasis Therapeutics Proteostasis
Therapeutics is developing disease-modifying therapeutics for cystic fibrosis, genetic diseases and neurodegenerative diseases. The Company's technology combines both phenotypic and target based drug discovery to develop therapeutics that modulate protein homeostasis pathways and correct for imbalances in protein folding, trafficking, and clearance. For more information, please visit www.proteostasis.com.
Investor Contact:
Malini Chatterjee, PhD
Blueprint Life Science Group
mchatterjee@bplifescience.com
(917) 330-4269
Cambridge, Mass. and Tokyo, Japan, November 4th, 2014 -- Proteostasis Therapeutics, Inc. (“Proteostasis Therapeutics”), a company developing novel therapeutics to address diseases caused by defects in protein folding, trafficking and clearance, and Astellas Pharma Inc. (“Astellas”) today announced that they have entered into a worldwide collaboration to research and develop therapeutic candidates that modulate the Unfolded Protein Response (UPR) through the use of Proteostasis Therapeutics’ proprietary “Disease Relevant Translation” (DRT™) and “Proteostasis Network” platform. Proteostasis Therapeutics will receive an initial upfront payment from Astellas, along with a securities investment. Proteostasis Therapeutics is also eligible for research funding support, future development and commercial milestones that could result in total payments of over $400 million, as well as tiered royalties. Further, Astellas has the right to begin two additional projects under the same terms, which, if it fully exercised this right, would bring the total potential value of the collaboration to $1.2B.
“We are very pleased to be collaborating with Astellas, a worldwide leader in the development of innovative therapeutics,” said Meenu Chhabra, President and CEO at Proteostasis Therapeutics. “Our novel approach to drug discovery, coupled with Astellas’ track record in drug development, will enable rapid discovery and development of therapies for important unmet medical needs.”
The collaboration will focus on one genetic disease, and further explore additional indications that can be affected through modulation of the UPR pathway. Stress induced by accumulation of unfolded proteins in the endoplasmic reticulum (ER) is observed in many diseases which are now recognized as protein conformational diseases, including genetic diseases, neurodegenerative diseases, and retinal degenerative diseases. Selective modulation of the UPR pathway in non-clinical investigations improved the stress response and restored function, suggesting that it can be beneficial as potential novel disease-modifying therapies for multiple diseases with high unmet medical needs.
“Proteostasis Therapeutics has a novel platform that offers a differentiated approach to discovering drugs for unmet medical needs,” commented Kenji Yasukawa, Ph.D., Senior Vice President and Chief Strategy Officer, at Astellas, “it complements our existing internal and externalized R&D initiatives and we continue to invest in innovative technologies that can provide new therapeutic options to patients. We look forward to working closely with Proteostasis Therapeutics to identify lead candidates for clinical development and potential commercialization.”
As part of the agreement, the Companies will conduct discovery, screening and preclinical research to identify lead compounds for clinical development. Upon candidate selection, Proteostasis Therapeutics will have the rights to opt in for global co-development and United States co-promotion.
This collaboration is being led by Innovation Management (“AIM”) of Astellas Pharma Inc.
The impact of this collaboration has been accounted in revised Astellas’ forecasts for fiscal year ending March 2015, which was announced on October 31st, 2014.
About Proteostasis Therapeutics, Inc.
Proteostasis Therapeutics, Inc. is a drug discovery company addressing diseases caused by defects in protein folding, trafficking and clearance for orphan and neurodegenerative diseases. The Company’s Disease Relevant Translation (DRT™) platform combines a proprietary screening approach with state-of-the-art medicinal chemistry, to generate highly selective drug candidates and is advancing a pipeline inclusive of a lead program in cystic fibrosis and a partnership with Biogen Idec for neurodegenerative diseases.
The Company’s DRT™ platform utilizes functionally pertinent assays and disease relevant models to identify highly translatable therapies associated with the modulation of protein homeostasis pathways within the cell. These pathways are part of cellular ‘quality control’ machinery, known as the protein homeostasis network or Proteostasis Network (PN). By modifying the function and capacity of the PN, the Company’s therapeutic product candidates correct for imbalances in the PN resulting from the cumulative effects of disease, genetic mutations, environmental factors and aging. For more information visit www.proteostasis.com.
About Astellas Pharma Inc.
Astellas Pharma Inc., located in Tokyo, Japan, is a pharmaceutical company dedicated to improving the health of people around the world through the provision of innovative and reliable pharmaceuticals. Astellas has approximately 18,000 employees worldwide. The organization is committed to becoming a global category leader in Urology, Immunology (including Transplantation) and Infectious diseases, Oncology, Neuroscience and Diabetes Mellitus (DM) Complications and Kidney diseases. For more information on Astellas Pharma Inc., please visit the company website at www.astellas.com/en.
About Innovation Management
Innovation Management ("AIM") is a new division established in October 2013 to enhance and accelerate the process of screening and realizing external opportunities to strengthen innovation at the preclinical development stage. AIM oversees strategic alliance activities with external partners and is responsible for a series of activities at acquiring external innovation opportunities in the preclinical development stage, such as strategy planning, screening, scientific assessment and alliance negotiations, so that strategic external business alliances can be strategically and systematically performed.
Investor Contacts:
Susan Shepard
Proteostasis Therapeutics, Inc.
sshepard@proteostasis.com
(617) 225-0096 x2113
Malini Chatterjee
Blueprint Life Science Group
mchatterjee@bplifescience.com
(917) 330-4269
Astellas Contacts:
Astellas Pharma Inc. Corporate Communications, +81-3-3244-3201
###
- Unum has built a universal cellular immunotherapy platform based on proprietary antibody-coupled T-cell receptor (ACTR) technology
CAMBRIDGE, MA, October 21, 2014 – Unum Therapeutics, a company developing cellular immunotherapy to treat cancer, announced today that it has raised $12 million in Series A funding led by Fidelity Biosciences and Atlas Venture, with participation from Sanofi-Genzyme BioVentures. Unum has built a platform for cancer treatment based upon an antibody-coupled T-cell receptor (ACTR). Genetically programming T-cells with ACTR allows them to efficiently attack and kill tumor cells using targeting antibodies. In contrast to other approaches that are limited to a single target and treat a narrow set of tumors, Unum’s approach is not restricted by antigen and may have applications for treating many types of cancers. Unum plans to use capital raised through this financing to advance its lead candidate through initial proof-of-concept clinical studies, further enhance the ACTR technology, and establish partnerships to access tumor-specific antibodies for a pipeline of novel combination therapies.
Industry veteran Charles “Chuck” Wilson, PhD, will serve as President & CEO. Before founding Unum, Dr. Wilson led the team responsible for partnering to support research and early development at Novartis. With many oncology and immuno-oncology deals to his credit, he brings extensive experience to Unum spanning from early drug discovery through Phase II clinical development. Prior to Novartis, Dr. Wilson held both scientific and business management roles in biotech.
“We’ve created Unum to rapidly develop this universal cell therapy platform and to explore its potential in a number of different cancer types,” said Dr. Wilson. “With our Series A funding from Fidelity Biosciences, Atlas Venture, and Sanofi-Genzyme BioVentures, we will drive our lead program into Phase I testing and aim to quickly validate the ACTR approach in the clinic.”
Dario Campana, MD, PhD, is the company’s Scientific Founder. Dr. Campana is an established leader in the field of cancer cell therapies. At the National University of Singapore (NUS), he developed the ACTR technology that forms the basis for Unum. At St. Jude Children’s Research Hospital (Memphis, TN), he created a chimeric antigen receptor (CAR) approach that is currently being pursued by several pharma and biotech companies. He currently oversees a number of cell therapy clinical trials in oncology.
In addition to Drs. Wilson and Campana, Unum Therapeutics’ management team also includes Chief Scientific Officer Seth Ettenberg, PhD, a cancer biologist and drug development scientist with extensive experience leading teams in biotechnology and large pharmaceutical drug discovery settings. Dr. Ettenberg most recently served as the Cambridge site head for Novartis Oncology Biotherapeutics.
The initial Board of Directors for Unum Therapeutics is comprised of:
- Chuck Wilson, PhD, President & CEO, Unum Therapeutics
- Ben Auspitz, Partner, Fidelity Biosciences
- Bruce Booth, DPhil, Partner, Atlas Venture
“Unum combines a strong, experienced management team, a transformational technology that may revolutionize cancer treatment, and top scientific and business development talent,” said Mr. Auspitz. “It has the right combination of resources to carry out its vision of bringing to market a single therapy that can augment the activity of many different antibodies to treat many different cancers.”
About Unum Therapeutics
Unum Therapeutics uses proprietary T-cell engineering technology in combination with tumor-targeting antibodies to activate the body’s own immune system to fight cancer. Unum’s lead program, based on its Antibody-Coupled T-cell Receptor (ACTR) technology, is expected to enter Phase I clinical testing in the coming months to assess safety and efficacy. Unum is seeking partners interested in using the ACTR technology to arm proprietary tumor-specific antibodies with a T-cell to improve their therapeutic potential. The company is headquartered in Cambridge, MA. For more information, visit www.unumrx.com.
About Fidelity Biosciences
Fidelity Biosciences (www.fidelitybiosciences.com) invests venture capital in biopharmaceutical, medical technology, healthcare information technology and healthcare service companies. The firm is a subsidiary of FMR LLC, the parent company of Fidelity Investments, one of the world’s leading providers of financial services. For more than 40 years, Fidelity has been a significant presence in the venture capital and private equity industry.
About Atlas Venture
Based in Cambridge, Massachusetts, Atlas Venture is focused on becoming the reference early-stage investor in New England. We are contrarian, patient, and aggressive investors. More information is available at www.atlasventure.com.
About Sanofi-Genzyme BioVentures
Sanofi-Genzyme BioVentures (SGBV) is a strategic fund that invests in early stage companies developing promising new products based on breakthrough science that may become future Sanofi pipeline candidates. SGBV’s investments align with Sanofi’s current and future areas of business interest such as rare diseases, oncology, vaccines, immune-mediated diseases, and other breakthrough therapies as well as integrated care solutions. For more information, visit us at www.sanofigenzymebioventures.com.
Media Contact:
Mike Beyer
Sam Brown Inc.
312.961.2502
beyer@sambrown.com
Cambridge, Mass., October 20, 2014 -- Proteostasis Therapeutics, Inc., a company developing novel therapeutics to address diseases caused by defects in protein folding, trafficking and clearance, today announced the appointment of six of the world’s leading cystic fibrosis experts to form its clinical advisory board. The newly appointed board will serve as a strategic resource for the upcoming selection and study of the Company’s leading compounds that double the activity of the most effective combination, ivacaftor/lumacaftor in the gold-standard HBE cell assay, for the most common mutation, F508del/F508del, found in the cystic fibrosis population.
“We are honored that these world renowned cystic fibrosis clinicians have agreed to bring their expertise to form our team of clinical advisors,” said Meenu Chhabra, President and Chief Executive Officer of Proteostasis Therapeutics. “The knowledge and experience that our inaugural clinical advisory board members bring to Proteostasis will be invaluable not only in the selection process of which of our proprietary PTI CFTR compounds should proceed to clinical development, but also in providing guidance on the design and conduct of the future clinical trials of selected drug candidates.”
Dr. Richard B. Moss, MD, a Professor Emeritus of Pediatrics at the Lucile Salter Packard Children’s Hospital at Stanford University, will serve as Chairman of the Clinical Advisory Board. He is the former Chief of Pediatric Pulmonary and Allergy Divisions, and allergy-immunology and pulmonary fellowship director. Dr. Moss also served as Director of the Cystic Fibrosis Center at Stanford, and site principal investigator for Cystic Fibrosis Therapeutics Development Network, where he was also the inaugural Chair of the Protocol Review Committee from 1991 to 2009.
Dr. Jane C. Davies, MD, FRCPCH, is Professor of Paediatric Respirology and Experimental Medicine at Imperial College London and an Honorary Consultant in Paediatric Respiratory Medicine at the Royal Brompton & Harefield NHS Foundation Trust. She is the site co-PI for the European CF Society Clinical Trials Network and leads their Lung Clearance Index Core facility. She has experience in clinical trial design for both investigator-initiated and pharma-sponsored studies.
Dr. Michael R. Knowles, MD, is Professor of Pulmonary and Critical Care Medicine at University of North Carolina (UNC), Chapel Hill. He is also the head of two multicenter studies, the first, Genetic modifiers of disease phenotype (severity) in cystic fibrosis lung and liver disease, which also includes a recently formed international consortium that is conducting a whole genome scan. The second is a consortium of eight North American sites studying rare genetic disorders of mucociliary clearance. Additionally, Dr. Knowles was the site PI at UNC Chapel Hill, and served on the Steering Committee, when the Cystic Fibrosis Therapeutic Development Network was founded.
Dr. Felix A. Ratjen, MD, PhD, is the Chief of Paediatric Respiratory Medicine at The Hospital for Sick Children, Professor of Paediatrics at The University of Toronto, and Senior Scientist at the Research Institute in the Department of Physiology and Experimental Medicine. He co-leads the Cystic Fibrosis Centre at The Hospital for Sick Children, and is the Medical Director of the Clinical Research Unit there.
Dr. Isabelle Sermet-Gaudelus, MD, PhD, is Professor of Pediatric Medicine at l'Hôpital Necker-Enfants Malades in Paris, France. She has developed and conducted several therapeutic trials for cystic fibrosis for both academic and company-initiated investigations. Her research focuses on modulation of the clinical severity of cystic fibrosis depending on fluid transfer and its therapeutic applications.
Dr. Pamela L. Zeitlin, MD, PhD, is Professor and Director of Pediatric Pulmonary Medicine and the Co-Director of the Cystic Fibrosis Center at Johns Hopkins University. Her research focuses on the role of chloride channels in inherited diseases of the respiratory tract, especially cystic fibrosis.
“As a pediatric pulmonologist, my research and clinical practice have been dedicated to helping children exercise one of their most basic rights, to breathe. The majority of my work has focused on finding the most up-to-date and effective treatments for children with cystic fibrosis, as they are still one of the few patient populations who are denied this right from birth, and have a life expectancy of 40. When Proteostasis asked me to lead their clinical advisory board, I was not only eager to learn all I could about their proprietary screening and drug development platform, but excited at the prospect of working with other clinicians who will bring their knowledge and experience of cystic fibrosis to an executive team making great strides in our field,” commented Dr. Moss, inaugural chairman of the clinical advisory board.
“The appointment of Proteostasis' clinical advisory board confirms our corporate commitment to developing innovative therapeutics for orphan diseases such as cystic fibrosis,” states Dr. Markus Haeberlein, PhD, Senior Vice President and Chief Scientific Officer at Proteostasis. “Our platform technology focuses on developing small molecules for protein folding trafficking and clearance diseases with few or no treatment options. Our proprietary screening and drug discovery process is broadly applicable to a number of diseases, and under the guidance of this advisory board, we are in an even better position to realize its true potential, starting with the cystic fibrosis patient population.”
About Proteostasis Therapeutics
Proteostasis is a drug discovery company addressing diseases caused by defects in protein folding, trafficking and clearance for orphan and neurodegenerative diseases. The Company’s Disease Relevant Translation (DRT™) platform combines a proprietary screening approach with state-of-the-art medicinal chemistry, to generate highly selective drug candidates and is advancing a pipeline inclusive of a lead program in cystic fibrosis and a partnership with Biogen Idec for neurodegenerative diseases.
The Company’s DRT™ platform utilizes functionally disease relevant cellular models with high translatability to develop therapies based on the modulation of protein homeostasis pathways within the cell. These pathways are part of cellular ‘quality control’ machinery, known as the protein homeostasis network or Proteostasis Network (PN). By modifying the function and capacity of the PN, the Company’s therapeutic product candidates correct for imbalances in the PN resulting from the cumulative effects of disease, genetic mutations, environmental factors and aging. For more information visit www.proteostasis.com.
Investor Contacts:
Susan Shepard Malini Chatterjee
Proteostasis Therapeutics, Inc. Blueprint Life Science Group
sshepard@proteostasis.com mchatterjee@bplifescience.com
(617) 225-0096 x2113 (917) 330-4269
SEATTLE and SOUTH SAN FRANCISCO – October 16, 2014 Immune Design Corp. (NASDAQ: IMDZ), a clinical-stage immunotherapy company, today announced that it has entered into a broad collaboration for the development of a herpes simplex virus (HSV) immune therapy with Sanofi Pasteur, the vaccines division of Sanofi (EURONEXT: SAN and NYSE: SNY).
Sanofi Pasteur and Immune Design will each contribute product candidates to the collaboration: Sanofi Pasteur will contribute HSV-529, a clinical-stage replication-defective HSV vaccine product candidate, and Immune Design will contribute G103, its preclinical trivalent vaccine product candidate. The collaboration will explore the potential of various combinations of agents, including leveraging Immune Design’s GLAASTM platform, with the goal to select the best potential immune therapy for patients.
The two companies will develop the products jointly through Phase 2 clinical trials, at which point Sanofi Pasteur intends to continue development of the most promising candidate and be responsible for commercialization. Sanofi Pasteur will bear the costs of all preclinical and clinical development, with Immune Design providing a specific formulation of GLA from the GLAAS platform at its cost through Phase 2 studies. Immune Design will be eligible to receive future milestone and royalty payments on any product developed from the collaboration; other financial terms of the agreement have not been disclosed.
“Instead of being limited to a single approach, the companies are joining forces and combining multiple cutting-edge technologies with the goal to develop the most effective and safe immunotherapy to address HSV infection, a significant unmet medical need,” said Carlos Paya, M.D., Ph.D., President and Chief Executive Officer of Immune Design. “With other clinical and preclinical GLAAS-based product candidates in development, both with partners and internally at Immune Design, I believe this new collaboration continues to demonstrate the productivity and broad applicability of this platform.”
About G103 and GLAAS
G103 is a trivalent vaccine candidate consisting of recombinantly-expressed viral proteins adjuvanted with a specific formulation from Immune Design’s GLAAS platform. The combination of a novel molecular toll-like receptor 4 (TLR4) agonist with rationally selected antigens is designed to boost pre-existing T cells and trigger a broad antibody response, allowing for prophylactic and therapeutic immunization.
The GLAAS platform works in vivo and is based on a small synthetic molecule called GLA, which stands for glucopyranosyl lipid adjuvant. GLA selectively binds to the TLR4 receptor and causes potent activation of dendritic cells (DCs) leading to the production of cytokines and chemokines that drive a Th1-type immune response. When GLA is accompanied by an antigen and injected into a patient, the combination is taken up by DCs and leads to the production and expansion of immune cells called CD4 T helper lymphocytes with a Th1 phenotype. These CD4 T cells play a key role in boosting pre-existing cytotoxic T cells that are specific to the same antigen; and providing help to other immune cells, including B lymphocytes that are the precursor to antibodies, and natural killer cells that are also important in the overall immune response. Immune Design believes that GLAAS- based product candidates have the potential to target multiple types of cancer, as well as infectious, allergic and autoimmune diseases. Product candidates leveraging GLAAS’ core technology have now
been evaluated in over 1000 subjects in Phase 1 and Phase 2 trials.
About Immune Design
Immune Design (NASDAQ: IMDZ) is a clinical-stage immunotherapy company employing next- generation in vivo approaches to enable the body’s immune system to fight disease. The company’s technologies are engineered to activate the immune system’s natural ability to create and/or expand antigen-specific cytotoxic T cells, while enhancing other immune effectors, to fight cancer and other chronic diseases. Immune Design’s three on-going immuno-oncology clinical programs are the product of its two synergistic discovery platforms: ZVexTMand GLAASTM, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute (IDRI), respectively. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
Immune Design Cautionary Note Regarding Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend", “believe” and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design’s expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include statements regarding efforts to develop products under the collaboration, the potential receipt of milestone and royalty payments and the potential to develop new therapeutics. Factors that may cause actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design’s filings with the U.S. Securities and Exchange Commission (the “SEC”), including the "Risk Factors" section of Immune Design’s Quarterly Report on Form 10-Q filed with the SEC on September 8, 2014 and in any subsequent filings with the SEC. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
# # #
For Immune Design:
Media Contact Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact Robert H. Uhl
Westwicke Partners
robert.uhl@westwicke.com
858-356-5932
NOVATO, Calif., Oct. 13, 2014 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (Nasdaq:RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced the presentation of results from a Phase 2 extension study of sialic acid extended-release (SA-ER, UX001) tablets in patients with hereditary inclusion body myopathy (HIBM; also known as GNE myopathy), a rare, progressive muscle-wasting disease. SA-ER is designed to replace the deficient sialic acid substrate in patients with HIBM. The data were presented at the 19th International Congress of the World Muscle Society (WMS) in Berlin.
"The data from the extension study of SA-ER support our plans to move forward with this program," said Sunil Agarwal, M.D. Chief Medical Officer of Ultragenyx. "While the 12 gram per day dose did not appear to have a clear advantage over the 6 gram per day dose, it does provide additional evidence of activity and safety and we are encouraged to see a potential long-term impact on disease progression in upper extremity muscle strength after approximately two years of treatment."
Patients in the initial Phase 2 study were randomized to receive placebo, 3 grams/day, or 6 grams/day of SA-ER. After 24 weeks, placebo patients crossed over to either 3 grams/day or 6 grams/day, on a blinded basis, for an additional 24 weeks. The 48-week analysis compared change from baseline for the combined groups at 6 grams/day versus 3 grams/day of SA-ER.
The initial Phase 2 data, which were presented at the American Academy of Neurology (AAN) Annual Meeting in April 2014, showed a statistically significant difference in the upper extremity composite (UEC) of muscle strength at 48 weeks with the higher dose group compared to the lower dose group. SA-ER appeared to be safe and well-tolerated with no serious adverse events observed to date. Most adverse events were mild to moderate and most commonly gastrointestinal in nature.
In the first part of the extension study, all 46 patients from the 48-week Phase 2 study crossed over to 6 grams/day for a variable period of time that was on average 24 weeks. In the second part of the extension study, all 46 patients and 13 treatment-naïve patients received 12 grams/day of SA-ER for 24 weeks. The results presented at WMS include the 49 out of 59 patients who had 24 weeks of data at the higher dose. While the 12 grams/day data do not suggest any clinically meaningful advantage over 6 grams/day, the 12 gram data do provide additional data that support clinical activity with SA-ER treatment. The 12 gram daily dose of SA-ER appeared to be generally safe and well tolerated with no drug-related serious adverse events, but the rate of mild to moderate gastrointestinal adverse events did appear to be greater with this dose. Over the entire approximate two-year study, treatment with SA-ER appeared to slow the progression of upper extremity disease when compared to the 24-week placebo group extrapolated out to two years.
Based on the 48-week and extension study data, Ultragenyx intends to discuss with regulatory authorities a potential pivotal study of SA-ER in HIBM patients. The company will also continue to treat patients in the ongoing extension study.
About Hereditary Inclusion Body Myopathy
Hereditary inclusion body myopathy (HIBM) is also known as GNE myopathy. HIBM is a rare, severe, progressive, genetic neuromuscular disease caused by a defect in the biosynthetic pathway for sialic acid, with onset in the late teens or twenties. The body's failure to produce enough sialic acid causes muscles to slowly waste away and can lead to very severe disability, with patients typically becoming wheelchair bound and losing most major muscle function within ten to 20 years from onset. There are approximately 1,200 to 2,000 HIBM patients in the developed world, and there is currently no approved therapy.
About Ultragenyx
Ultragenyx is a clinical-stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with a focus on serious, debilitating genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
Forward-Looking Statements
Except for the historical information contained herein, the matters set forth in this press release, including statements regarding the potential impact of SA-ER on the progression of HIBM and plans for a potential pivotal study, are forward-looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance, or achievements to differ significantly from those expressed or implied by the forward-looking statements. Such risks and uncertainties include, among others, the uncertainties inherent in the clinical drug development process, including the regulatory approval process, the timing of our regulatory filings, and other matters that could affect the availability or commercial potential of our drug candidate. Ultragenyx undertakes no obligation to update or revise any forwardlooking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on August 11, 2014, and its subsequent periodic reports filed with the Securities and Exchange Commission.
CONTACT:
Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
SEATTLE and SOUTH SAN FRANCISCO, Calif., Oct. 2, 2014 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ) announced today that the company was added to the Russell 2000® Index as part of Russell Investments' quarterly addition of select initial public offering (IPO) companies. Immune Design joined the index after the NASDAQ market closed on September 30, 2014. The stock also was added systematically to the appropriate Russell growth and value indexes.
The Russell 2000 Index measures the performance of the small-cap segment of the U.S. equity universe and is a subset of the Russell 3000® Index. Russell indexes are used by investment managers and institutional investors for index funds and as benchmarks for active investment strategies.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to create and/or expand antigen-specific cytotoxic T cells, while enhancing other immune effectors, to fight cancer and other chronic diseases. Immune Design's three on-going Immuno-oncology clinical programs are the product of its two synergistic discovery platforms: ZVexTMand GLAASTM. Immune Design has offices in Seattle and South San Francisco. For more information, visit www.immunedesign.com.
CONTACT:
Company Contact
Stephen R. Brady
Chief Business Officer
650-887-6717
Media Contact
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
Investor Contact
Robert H. Uhl
Westwicke Partners
robert.uhl@westwicke.com
858-356-5932
All Patients Continued to Demonstrate Increases in Serum Phosphorus and the Majority Maintained Levels in the Normal Range
NOVATO, Calif., Sept. 15, 2014 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (Nasdaq:RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced the presentation of results from a long-term Phase 1/2 extension study, conducted by Kyowa Hakko Kirin Pharma, Inc., of the investigational fully human anti-FGF23 monoclonal antibody KRN23 (UX023) in adult patients with X-linked hypophosphatemia (XLH). The cumulative 16-month data, combining the four-month dose escalation period data from INT-001 and the 12-month extension data from INT-002, were presented at the American Society of Bone and Mineral Research (ASBMR) Annual Meeting in Houston.
"By binding and inhibiting FGF23, patients treated with KRN23 demonstrated increases in phosphate levels over the cumulative 16-month treatment period," said Sunil Agarwal, M.D., Chief Medical Officer of Ultragenyx. "Based on these encouraging results, we plan to continue development in adult XLH patients and are enrolling pediatric XLH patients in our ongoing Phase 2 study."
The Phase 1/2 extension study (INT-002) was designed to evaluate long-term safety and efficacy following an initial four-month dose escalation study (INT-001) that was conducted in the US and Canada. During the extension study, 22 adult patients with XLH were evaluated over an additional 12 months. Patients received monthly subcutaneous injections of KRN23 administered at a dose range of 0.1 to 1.0 mg/kg. Data from the INT-001 study were previously presented at the ICE/ENDO joint meeting of The Endocrine Society and The International Congress of Endocrinology in June 2014.
Data from the INT-002 study demonstrated that the increases in serum phosphorus levels, urinary phosphorus reabsorption, and 1,25 dihydroxy vitamin D levels observed in the initial INT-001 study were generally sustained during the 12-month extension. All patients continued to demonstrate increases in serum phosphorus levels. Approximately 52.6%-85.7% of subjects in the extension study had serum phosphorus levels that reached the normal range (2.5 to 4.5 mg/dL) at peak time on Day 7 or Day 14 after each dose over this 12-month period.
The mean increases in markers of bone remodeling (procollagen type I N propeptide (P1NP) and osteocalcin) observed in INT- 001 were also generally sustained.
KRN23 was generally safe and well tolerated over the cumulative treatment period. The most common treatment-related adverse events were injection site reaction, arthralgia, diarrhea, restless legs syndrome, injection site erythema, injection site pain, upper abdominal pain, headache, and decreased neutrophil count (the neutrophil changes were not associated with any significant infections). Serious adverse events were reported in three subjects but were all considered unrelated to KRN23. One patient discontinued treatment due to nephrolithiasis and one patient discontinued due to restless legs syndrome. There were no significant changes in parathyroid hormone or renal ultrasound. Serum calcium levels did not change significantly, and mild hypercalcemia was observed intermittently in two subjects. Urinary calcium was not increased, and three subjects had only transient hypercalciuria. No anti-KRN23 antibodies were observed.
Ultragenyx and Kyowa Hakko Kirin Co., Ltd. (Tokyo:4151) initiated a Phase 2 study of KRN23 in pediatric patients in the US and EU in June 2014 and expect to continue the clinical development of KRN23 in adults with XLH.
About X-Linked Hypophosphatemia (XLH)
XLH is a disorder of phosphate metabolism caused by phosphate wasting in the urine leading to severe hypophosphatemia. XLH is the most common heritable form of rickets that is inherited as an X-linked dominant trait affecting both males and females, though some reports indicate that the disease may be more severe in males. Studies suggest there are approximately 12,000 XLH patients in the United States. XLH is a distinctive bone disease characterized by inadequate mineralization of bone that leads to a spectrum of abnormalities, including rickets, progressive bowing of the leg, osteomalacia, bone pain, waddling gait, short stature, gross motor impairment, muscle weakness, osteopenia, frequent/poorly healing microfractures, spinal stenosis, enthesopathy, and osteoarthritis.
Most patients are managed using oral phosphate replacement and vitamin D (calcitriol) therapy, which requires frequent divided doses and careful medical monitoring. It is partially effective at reducing rickets, but it does not improve growth and can be challenging to optimize the dose without increasing the risk of depositing phosphate-calcium precipitates in the kidneys (nephrocalcinosis).
About KRN23 and FGF23
KRN23 is an investigational recombinant fully human monoclonal IgG1 antibody, discovered by KHK, against the phosphaturic hormone fibroblast growth factor 23 (FGF23). It is being developed to treat XLH, a disease characterized by excess activity of FGF23. FGF23 is a hormone that reduces serum levels of phosphorus and vitamin D by regulating phosphate excretion and vitamin D production by the kidney. Phosphate wasting in XLH is caused by excessive levels and activity of FGF23. KRN23 is designed to bind to and thereby inhibit the excessive biological activity of FGF23. By blocking excess FGF23 in patients with XLH, KRN23 is intended to restore normal phosphate reabsorption from the kidney and increase the production of vitamin D, which enhances intestinal absorption of phosphate and calcium. Ultragenyx and KHK entered into a collaboration and license agreement in August 2013 to develop and commercialize KRN23.
About Ultragenyx
Ultragenyx is a clinical-stage biotechnology company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with a focus on serious, debilitating genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
About Kyowa Hakko Kirin
Kyowa Hakko Kirin Co., Ltd. (KHK) is a leading biopharmaceutical company in Japan focusing on its core business area of oncology, nephrology, and immunology/allergy. KHK leverages antibody-related leading-edge technologies to discover and develop innovative new drugs aiming to become a global specialty pharmaceutical company which contributes to the health and well-being of people around the world. Kyowa Hakko Kirin Pharma, Inc. is a subsidiary of KHK.
For more information, please visit http://www.kyowa-kirin.com.
Forward-Looking Statements
Except for the historical information contained herein, the matters set forth in this press release, including statements regarding the plans for continued development of KRN23 in adult and pediatric patients, are forward-looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance, or achievements to differ significantly from those expressed or implied by the forward-looking statements. Such risks and uncertainties include, among others, the uncertainties inherent in the clinical drug development process, including the regulatory approval process, the timing of our regulatory filings, that these results (which were in adult patients) may not translate into similar safety or efficacy in pediatric patients, and other matters that could affect the availability or commercial potential of our drug candidates. Ultragenyx undertakes no obligation to update or revise any forward-looking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on May 12, 2014, and its subsequent periodic reports filed with the Securities and Exchange Commission.
CONTACT:
Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
Rapid and Sustained Reduction in Urinary Substrate Excretion Observed Over 12 Weeks
NOVATO, Calif., Sept. 3, 2014 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (Nasdaq:RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced the presentation of positive interim data from the Phase 1/2 study of recombinant human beta-glucuronidase (rhGUS, UX003), an investigational therapy for the treatment of mucopolysaccharidosis 7 (MPS 7, Sly syndrome). The data are being presented at the Society for the Study of Inborn Errors of Metabolism (SSIEM) Annual Symposium in Innsbruck, Austria.
The Phase 1/2 open-label clinical study is assessing the safety, efficacy, and dose of rhGUS administered every other week via intravenous infusion in three patients. A 12-week primary analysis phase evaluating 2 mg/kg of rhGUS every other week is being followed by dose-exploration and long-term extension.
"We are pleased with the 12-week results of the first clinical study to be conducted in MPS 7 and are grateful to the patients and investigators who are participating in the study," commented Emil D. Kakkis, Ph.D., M.D., Chief Executive Officer and President of Ultragenyx. "Based on the reduction of lysosomal storage shown in all patients in the Phase 1/2 study, we plan to move into Phase 3 testing."
Results from the primary analysis phase show evidence of clearance of lysosomal storage as indicated by the decline in urinary glycosaminoglycan (GAG) excretion and the reduction in liver size. The change in urinary GAG excretion was observed by two weeks after the first dose of rhGUS and declined by approximately 40-50% from baseline after 12 weeks of treatment. Decreases in liver size were observed in the two patients who had enlarged livers at baseline.
No serious adverse events were observed in the 12-week primary analysis phase and through up to 28 total weeks of treatment. The most common adverse events reported to date are infections and gastrointestinal disorders. No infusionassociated reactions were observed after a total of 38 infusions to date in these three subjects.
In addition to the Phase 1/2 study, one patient continues to be treated under an emergency Investigational New Drug application (eIND) sponsored by Dr. Joyce Fox and the Steven and Alexandra Cohen Children's Medical Center of New York. Through 24 weeks of treatment, a decline in urinary GAG excretion of 50-70% and a sustained reduction in the size of the enlarged liver and spleen have been observed. The data also show improved pulmonary function based on reduced carbon dioxide retention. No serious adverse events or infusion-associated reactions were observed through 12 infusions.
The Phase 1/2 study will continue through the dose-exploration phase and long-term extension. Based on the Phase 1/2 study results and the 24-week results of the patient treated under an eIND, the company intends to initiate a pivotal Phase 3 study by year-end 2014.
About MPS 7
Mucopolysaccharidosis type 7 (MPS 7, Sly syndrome), originally described in 1973 by William Sly, M.D., is a rare genetic, metabolic disorder and is one of 11 different MPS disorders. MPS 7 is caused by the deficiency of beta-glucuronidase, an enzyme required for the breakdown of the glycosaminoglycans (GAGs) dermatan sulfate and heparan sulfate. These complex GAG carbohydrates are a critical component of many tissues. The inability to properly break down GAGs leads to a progressive accumulation in many tissues and results in a multi-system disease.
While its clinical manifestations are similar to MPS 1 and MPS 2, MPS 7 is one of the rarest among the MPS disorders. MPS 7 has a wide spectrum of clinical manifestations and can present as early as at birth. There are no approved therapies for MPS 7 today. The use of enzyme replacement therapy as a potential treatment is based on 20 years of research work in murine models of the disease. Enzyme replacement as a strategy is well established in the MPS field as there are currently four approved enzyme replacement therapies for other MPS disorders: MPS 1 (Aldurazyme®, laronidase), MPS 2 (Elaprase®, idursulfase), MPS 4A (Vimizim™, elosulfase alfa), and MPS 6 (Naglazym®e, galsulfase).
Ultragenyx initiated a Phase 1/2 study in the UK to evaluate the safety, tolerability, efficacy, and dose of intravenous administration of rhGUS in December 2013.
About Ultragenyx
Ultragenyx is a clinical-stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with a focus on serious, debilitating metabolic genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
Forward-Looking Statements
Except for the historical information contained herein, the matters set forth in this press release, including statements regarding Ultragenyx's expectations regarding the timing of release of data, plans regarding continuation of the Phase ½ study and the timing of initiation of a pivotal Phase 3 study, are forward-looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance, or achievements to differ significantly from those expressed or implied by the forward-looking statements. Such risks and uncertainties include, among others, the uncertainties inherent in the clinical drug development process, including the regulatory approval process, the timing of our regulatory filings, and other matters that could affect the availability or commercial potential of our drug candidate. Ultragenyx undertakes no obligation to update or revise any forward-looking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on May 12, 2014, and its subsequent periodic reports filed with the Securities and Exchange Commission.
CONTACT:
Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
-- Anti-EphA3 Agents Inhibit Tumor Growth by Disrupting or Destroying the Tumor Microenvironment:
Malignant Stem Cells, Stromal Cells, and New Tumor Vasculature --
SOUTH SAN FRANCISCO, Calif., Aug. 14, 2014 /PRNewswire/ -- KaloBios Pharmaceuticals, Inc. (Nasdaq: KBIO) today announced the publication of preclinical findings describing EphA3 as a novel target expressed by a broad range of human tumors, and whose activation can lead to the selective disruption of the tumor microenvironment and newly formed tumor blood vessels. The expression and function of EphA3 in the tumor microenvironment but not in adult normal tissues together with the favorable safety profile as observed thus far in a Phase 1-2 clinical study, where infusion reactions have been the most common adverse event, supports the development of KB004 (KaloBios' Humaneered® anti-EphA3 monoclonal antibody [mAb]) as a potential treatment for both hematologic and solid tumors.
The new research findings, published online this week by the journal of the American Association for Cancer Research, are described by scientists from Monash University School of Biomedical Sciences and Ludwig Institute for Cancer Research in Melbourne, Australia, and KaloBios.
"Eph receptors constitute the largest family of receptor tyrosine kinases, and EphA3 is important in human fetal development. EphA3 is also found in the mesenchymal tissues of various developing organs but is virtually undetectable in normal adult tissues," said Professor Andrew Scott, from the Ludwig Institute for Cancer Research. "However, EphA3 is over-expressed in many solid and hematologic tumors where it is implicated in cell positioning and possible tumor stem cell survival."
He continued, "In this publication, we show that EphA3 is prominently expressed and functional in the tumor microenvironment, where it contributes to the formation of new blood vessels, and is also found on stromal tissue. We have also demonstrated that treatment of human tumor xenografts with a highly specific anti-EphA3 monoclonal antibody can effectively kill tumor-resident mesenchymal/stromal stem cells and inhibit tumor growth by disrupting the architecture and function of the tumor microenvironment."
"These data add to the rationale for our current clinical program using anti-EphA3 to treat leukemias and represent a clear rationale for the potential expansion of our investigation of KB004, KaloBios' Humaneered® anti-EphA3 mAb, into solid tumor indications," said Geoffrey Yarranton, Ph.D., KaloBios' chief scientific officer and executive vice president of research and development. KaloBios is currently conducting a Phase 1/2 clinical trial of KB004 in hematologic malignancies, with the Phase 2 expansion portion of that trial focused on acute myeloid leukemia (AML), myelodysplastic syndrome (MDS), and myelofibrosis (MF) patients with EphA3-positive malignancies.
"Research points to stromal cells playing a key role in chemoresistance, protecting tumors from the killing effects of many anticancer drugs. Thus, if KB004 is able to kill these cells protecting either solid tumors or tumors in the bone marrow, it might offer a particular powerful new way to fight such cancers," Dr. Yarranton noted.
About KaloBios
KaloBios Pharmaceuticals, Inc. is developing a portfolio of proprietary, patient-targeted, first-in-class monoclonal antibodies designed to treat severe life-threatening or debilitating diseases for which there is an unmet medical need, with a clinical focus on severe respiratory diseases and cancer.
Currently, KaloBios has advanced three programs to clinical development:
- l KB001-A is an anti-PcrV mAb fragment being developed for the prevention and treatment of Pseudomonas aeruginosa (Pa) infection. KaloBios is conducting a 180 patient Phase 2 study in cystic fibrosis (CF) subjects with chronic Pa lung infection. KaloBios has received Orphan Drug designation from both the U.S. FDA and the European Medicines Agency for KB001-A for the treatment of Pa lung infection in CF patients. KB001-A has also received Fast Track Status from the U.S. FDA for the prevention of ventilator associated pneumonia. KaloBios is planning to seek a partner to help accelerate the development of this program.
- l KB004 is an anti-EphA3 mAb with potential in treating hematologic malignancies and solid tumors. KaloBios is running an ongoing Phase 1/2 study evaluating KB004 in hematologic malignancies. The Phase 1 dose escalation portion of that study in subjects with hematologic malignancies is fully enrolled with dosing ongoing. KaloBios initiated the Phase 2 expansion portion of the study focused on patients with certain EphA3 positive hematologic malignancies in 2014.
- l KB003 is an anti-GM-CSF mAb with potential to treat inflammatory diseases that was being developed for the treatment of severe asthma. In early 2014, KaloBios completed a Phase 2 clinical study in 160 patients with severe asthma which did not meet its primary endpoint of improvement in FEV1 from baseline as compared to placebo. As a result, KaloBios discontinued development of this compound in severe asthma, and is continuing to analyze the Phase 2 data to review with thought leaders. KaloBios is currently evaluating other possible indications in order to determine next steps, if any, in the development of KB003.
All of the company's antibodies were generated using its proprietary Humaneered® technology, a method that converts nonhuman antibodies (typically mouse) into recombinant antibodies that have a high binding affinity to their target and are designed for chronic therapeutic use. The company believes that antibodies produced using its Humaneered® technology offer important clinical and economic advantages over antibodies generated by other methods in terms of high binding affinity, high manufacturing yields, and minimal to no immunogenicity (inappropriate immune response) upon repeat administration in humans.
For more information on KaloBios Pharmaceuticals, please visit our web site at http://www.kalobios.com.
Forward Looking Statements
This release contains forward-looking statements made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995, and statements regarding the company's clinical development of KB001-A, KB004 and KB003. Forward-looking statements reflect management's current knowledge, assumptions, judgment and expectations regarding future performance or events. Although management believes that the expectations reflected in such statements are reasonable, they give no assurance that such expectations will prove to be correct and you should be aware that actual results could differ materially from those contained in the forward-looking statements. Forward-looking statements are subject to a number of risks and uncertainties including, but not limited to, the potential timing and outcomes of clinical studies of KB001-A and KB004 undertaken now or in the future; the potential, if any, for future development of KB003; the company's limited cash reserves and its ability to obtain additional capital on acceptable terms, or at all, including the additional capital which will be necessary to complete the clinical trials that the company has initiated or plans to initiate; the company's dependence on Sanofi Pasteur for the manufacture, development and commercialization of KB001-A; the company's ability to successfully progress or complete further development of its programs; the uncertainties inherent in clinical testing; the timing, cost and uncertainty of obtaining regulatory approvals; the company's ability to protect the company's intellectual property; competition; changes in the regulatory landscape or the imposition of regulations that affect the company's products; and other factors listed under "Risk Factors" in the company's most recent quarterly report on Form 10-Q filed with the Securities and Exchange Commission on May 8, 2014, the Annual Report on Form 10-K filed on March 13, 2014, the quarterly reports on Form 10-Q filed on May 14, August 19, and November 12, 2013, and the company's other filings with the Securities and Exchange Commission.
All forward-looking statements are expressly qualified in their entirety by this cautionary notice. You are cautioned not to place undue reliance on any forward-looking statements, which speak only as of the date of this release. The company has no obligation, and expressly disclaims any obligation to update, revise or correct any of the forward-looking statements, whether as a result of new information, future events or otherwise.
For more information, visit http://www.kalobios.com.
Contact:
Herb C. Cross
Chief Financial Officer
KaloBios Pharmaceuticals, Inc.
(650) 243-3114
ir@kalobios.com
Media Contact:
Joan E. Kureczka
Kureczka/Martin Associates
Tel: (415) 821-2413
Mobile: (415) 690-0210
Joan@Kureczka-Martin.com
SOURCE KaloBios Pharmaceuticals, Inc.
News Provided by Acquire Media
CAMBRIDGE, Mass. and SEATTLE and SOUTH SAN FRANCISCO, Calif., Aug. 7, 2014 (GLOBE NEWSWIRE) -- Sanofi
(EURONEXT:SAN) (NYSE:SNY) and Immune Design (Nasdaq:IMDZ), a clinical-stage immunotherapy company, today announced that they have entered into a licensing agreement for use of Immune Design's GLAASTM discovery platform to develop therapeutic agents to treat a selected food allergy.
The incidence of food allergies is increasing worldwide in both developed and undeveloped countries, and especially in children.1 Globally, experts believe 220-250 million people may suffer from food allergies.2,3 In the United States alone, as many as 15 million people have food allergies,4 with allergic reactions resulting in an emergency room visit every three minutes and averaging more than 200,000 emergency room visits per year.5
"This is an exciting time in the area of immunology research, and our relationship with Immune Design is a great example of how Sanofi has changed our approach to R&D," said Kurt Stoeckli, vice president and head of Global Bio Therapeutics Organization, Sanofi. "With this partnership, we are able to tap into breakthrough science that holds great potential to transform how food allergies are treated, and the lives of those people affected. This kind of innovation is central to our new approach."
Under terms of the agreement, Immune Design has granted Sanofi an exclusive license to discover, develop and commercialize products to treat a selected food allergy. The company has received an undisclosed upfront payment and will be eligible to receive development and commercialization milestones totaling US $168 million, as well as tiered royalties on sales of approved products.
"Our fourth agreement for the use of the GLAAS platform further demonstrates the broad applicability of this approach not only in cancer and infectious diseases, but now in allergic diseases as well," said Stephen Brady, chief business officer at Immune Design. "Due to the immune dysfunction leading to allergic diseases, GLAAS' mechanism of action is well suited to correct the imbalance, allowing for the potential of new therapeutics in the targeted indication that currently uses century-old technologies. We are pleased that Sanofi has decided to develop products for this often life-threatening and growing food allergy."
Under an existing collaborative research arrangement, Sanofi and Immune Design have generated a large set of preclinical data demonstrating that certain formulations within GLAAS, when given prophylactically or therapeutically, can shift the immune responses in a way that may result in significant protection and reduction from allergy symptoms.
About Sanofi
Sanofi, an integrated global healthcare leader, discovers, develops and distributes therapeutic solutions focused on patients' needs. Sanofi has core strengths in the field of healthcare with seven growth platforms: diabetes solutions, human vaccines, innovative drugs, and consumer healthcare, emerging markets, animal health and the new Genzyme. Sanofi is listed in Paris (EURONEXT:SAN) and in New York (NYSE:SNY).
About GLAAS
Immune Design's GLAAS platform works in vivo and is based on a small synthetic molecule called GLA, which stands for glucopyranosyl lipid adjuvant. GLA selectively binds to the TLR4 receptor and causes potent activation of dendritic cells (DCs) leading to the production of cytokines and chemokines that drive a Th1-type immune response. When GLA is accompanied by an antigen and injected into a patient, the combination is taken up by DCs and leads to the production and expansion of immune cells called CD4 T helper lymphocytes with a Th1 phenotype. These CD4 T cells play a key role in boosting pre- existing CTLs that are specific to the same antigen; and providing help to other immune cells, including B lymphocytes that are the precursor to antibodies, and natural killer cells that are also important in the overall immune response. Immune Design believes that GLAAS product candidates have the potential to target multiple types of cancer, as well as infectious, allergic and autoimmune diseases. GLAAS-based product candidates have now been evaluated in over 1000 subjects in Phase 1 and Phase 2 trials demonstrating an acceptable safety profile and efficacy.
About Immune Design
Immune Design (Nasdaq:IMDZ) is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to create tumor-specific cytotoxic T cells, while enhancing other immune effectors, to fight cancer and other chronic diseases. Immune Design's three on-going Immuno-oncology clinical programs are the product of its two synergistic discovery platforms: DCVexTMand GLAASTM, the fundamental technologies of which were licensed from the California Institute of Technology and the Infectious Disease Research Institute, respectively. Immune Design has offices in Seattle, Washington and South San Francisco, California. For more information, visit www.immunedesign.com.
Sanofi Forward-Looking Statements
This press release contains forward-looking statements as defined in the Private Securities Litigation Reform Act of 1995, as amended. Forward-looking statements are statements that are not historical facts. These statements include projections and estimates and their underlying assumptions, statements regarding plans, objectives, intentions and expectations with respect to future financial results, events, operations, services, product development and potential, and statements regarding future performance. Forward-looking statements are generally identified by the words "expects", "anticipates", "believes", "intends", "estimates", "plans" and similar expressions. Although Sanofi's management believes that the expectations reflected in such forward-looking statements are reasonable, investors are cautioned that forward-looking information and statements are subject to various risks and uncertainties, many of which are difficult to predict and generally beyond the control of Sanofi, that could cause actual results and developments to differ materially from those expressed in, or implied or projected by, the forward-looking information and statements. These risks and uncertainties include among other things, the uncertainties inherent in research and development, future clinical data and analysis, including post marketing, decisions by regulatory authorities, such as the FDA or the EMA, regarding whether and when to approve any drug, device or biological application that may be filed for any such product candidates as well as their decisions regarding labelling and other matters that could affect the availability or commercial potential of such product candidates, the absence of guarantee that the product candidates if approved will be commercially successful, the future approval and commercial success of therapeutic alternatives, the Group's ability to benefit from external growth opportunities, trends in exchange rates and prevailing interest rates, the impact of cost containment policies and subsequent changes thereto, the average number of shares outstanding as well as those discussed or identified in the public filings with the SEC and the AMF made by Sanofi, including those listed under "Risk Factors" and "Cautionary Statement Regarding Forward-Looking Statements" in Sanofi's annual report on Form 20-F for the year ended December 31, 2013. Other than as required by applicable law, Sanofi does not undertake any obligation to update or revise any forward-looking information or statements.
Immune Design Cautionary Note Regarding Forward-Looking Statements
This press release contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend", "believe" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward-looking statements. These forward-looking statements are based on Immune Design's expectations and assumptions as of the date of this press release. Each of these forward-looking statements involves risks and uncertainties. Actual results may differ materially from these forward-looking statements. Forward-looking statements contained in this press release include statements regarding the receipt of milestone and royalty payments, the potential to develop new therapeutics and the potential of any future products to prevent and reduce allergy symptoms. Factors that may cause actual results to differ from those expressed or implied in the forward-looking statements in this press release are discussed in Immune Design's filings with the U.S. Securities and Exchange Commission, including the "Risk Factors" contained therein. Except as required by law, Immune Design assumes no obligation to update any forward-looking statements contained herein to reflect any change in expectations, even as new information becomes available.
References
- "Food Allergy - A Rising Global Health Problem," World Allergy Week 2013. 8-14 April 2013. http://www.worldallergy.org/UserFiles/file/WorldAllergyWeek2013final.pdf. Accessed online, July 28, 2014.
- Mills EN, Mackie AR, Burny P, Beyer K, Frewer L et al. "The prevalence, cost and basis of food allergy across Europe." Allergy 2007; 62:717-722.
- Fiocchi A, Sampson HA. "Food Allergy", Section 2.5, in WAO White Book on Allergy, Pawankar R, Canonica GW, Holgate ST, and Lockey RF, editors (Milwaukee, Wisconsin: World Allergy Organization, 2011), pp. 47-53.
- National Institute of Allergy and Infectious Diseases, National Institutes of Health. Report of the NIH Expert Panel on Food Allergy Research. 2006. Accessed online, July 25, 2014. http://www.niaid.nih.gov/topics/foodallergy/research/pages/reportfoodallergy.aspx
- 5. Clark S, Espinola J, Rudders SA, Banerji, A, Camargo CA. Frequency of US emergency department visits for food-
related acute allergic reactions. J Allergy ClinImmunol. 2011; 127(3):682-683.
CONTACT:
Amy BA, Ph.D.
Sanofi Global R&D Communications
Amy.Ba@sanofi.com
Tel: 646-207-4935
Julie Rathbun
Rathbun Communications (Immune Design)
julie@rathbuncomm.com
Tel: 206-769-9219
New Patent Also Issued With Composition Claims for Triheptanoin
NOVATO, Calif., Aug. 5, 2014 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (Nasdaq:RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced a license agreement with UniQuest Pty Limited for intellectual property related to the treatment of refractory epilepsy and other seizurerelated and neurologic disorders with triheptanoin (UX007). The intellectual property originated from research on epilepsy and other neurologic models conducted at The University of Queensland.
Separately, a United States (U.S.) patent has been issued with claims directed to compositions of triheptanoin above a certain level of purity. The patent term expires in October 2025. A second U.S. patent with claims directed to compositions of triheptanoin in a dosage unit was issued in August 2013 and expires in June 2020. These patent terms do not include any potential for patent term extension of up to five additional years. Similar claims are either granted or being pursued by Ultragenyx in other territories outside the U.S.
"The completion of the license agreement with UniQuest and the issuance of additional composition patent claims mark the continued strengthening of our triheptanoin portfolio," commented Emil D. Kakkis, Ph.D., M.D., Chief Executive Officer and President of Ultragenyx. "We are excited about the potential for triheptanoin to be used in certain difficult-to-treat seizure disorders and hope it will provide a new treatment option for affected patients."
About Refractory Epilepsy
Epilepsy is a family of chronic neurological disorders characterized by spontaneous recurrent seizures. There are
approximately two million epilepsy patients in the U.S. of various types, some of which are genetic in origin. Refractory epilepsy occurs in patients whose seizures cannot be adequately controlled by anti-epileptic drugs, which happens in approximately one third of cases.
About Triheptanoin
Triheptanoin, also known as UX007, is a purified form of a specially designed synthetic triglyceride compound. Ultragenyx is currently evaluating triheptanoin in two clinical programs.
The first program is studying the genetic seizure disorder Glut1 deficiency syndrome (Glut1 DS). This disease is caused by a genetic defect in the transport of glucose into the brain and affects an estimated 3,000 to 7,000 patients in the U.S. Glut1 DS is characterized by seizures, developmental delay, and movement disorder. Triheptanoin is metabolized to and intended to provide patients with heptanoate, which can diffuse across the blood-brain barrier and be converted into glucose. Heptanoate can also be further metabolized to four- and five-carbon ketone bodies in the liver that also cross the blood-brain-barrier and provide an additional energy source to the brain. Heptanoate and five-carbon ketone bodies can also regenerate new glucose in the brain, which is deficient in these patients. Ultragenyx is conducting a Phase 2 study in the U.S. and Europe to evaluate the potential of triheptanoin to treat Glut1 DS patients who have failed the ketogenic diet and who continue to have
breakthrough seizures.
The second program is studying long-chain fatty acid oxidation disorders (LC-FAOD), a set of rare metabolic diseases caused by the inability to convert fat into energy, leading to low blood sugar, muscle rupture, and heart and liver disease. Triheptanoin is intended to provide patients with medium-length, odd-chain fatty acids that are metabolized to increase intermediate substrates in the Krebs cycle, a key energy-generating process. Unlike typical even-chain fatty acids, triheptanoin can be converted to new glucose through the Krebs cycle, potentially providing an important added therapeutic effect, particularly when glucose levels are too low. Ultragenyx is conducting a Phase 2 study to evaluate the potential of triheptanoin to treat LCFAOD.
About Ultragenyx
Ultragenyx is a clinical-stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with a focus on serious, debilitating genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies. The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
Forward-Looking Statements
Except for the historical information contained herein, the matters set forth in this press release, including statements regarding any potential for patent term extension, the pursuit of patent claims in territories outside of the U.S., the potential for triheptanoin to be used in difficult-to-treat seizure disorders and to provide a new treatment option for seizure treatment of these patients and the potential for triheptanoin to provide a therapeutic effect to patients with LC-FAOD, are forward-looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance, or achievements to differ significantly from those expressed or implied by the forward-looking statements. Such risks and uncertainties include, among others, the uncertainties related to patent term extension, as well as uncertainties inherent in the clinical drug development process, including the regulatory approval process, the timing of our regulatory filings, and other matters that could affect the availability or commercial potential of our drug candidates. Ultragenyx undertakes no obligation to update or revise any forward-looking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on May 12, 2014, and its subsequent periodic reports filed with the Securities and Exchange Commission.
CONTACT:
Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
Strengthened Cash Position Expected to Fund Company Through 2015
SAN DIEGO, July 31, 2014 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE), a biopharmaceutical company engaged in the discovery and development of adult stem cell modulators to treat orphan diseases, announced today that it has completed a long-term debt financing of up to $20 million with Silicon Valley Bank. The Company has drawn down $10 million, at a fixed interest rate of 6.9%, under the first tranche of the debt facility.
"This debt financing further secures the necessary cash resources to clinically validate our lead product candidate PROHEMA® across multiple disease franchises including adult and pediatric patients with hematologic malignancies and pediatric patients with inherited metabolic disorders," said J. Scott Wolchko, Chief Financial & Operating Officer of Fate Therapeutics, Inc. "The additional capital also enables the Company to invest in, and achieve a number of important milestones in connection with, its pipeline of disease-altering hematopoietic- and muscle-based cellular therapeutics."
Fate Therapeutics is currently enrolling patients in its Phase 2 PUMA study, a randomized, controlled clinical trial that is designed to assess the efficacy and safety of PROHEMA® (16, 16-dimethyl prostaglandin E2, or dmPGE2, modulated cord blood) in adult patients undergoing hematopoietic stem cell transplantation (HSCT) for the treatment of hematologic malignancies. Safety reviews are planned after six and 12 subjects, respectively, have been treated with PROHEMA® in the PUMA study, and the Company intends to provide a clinical update following the completion of these reviews. Full data on the primary efficacy endpoint from the PUMA study are expected in mid-2015. In addition, Fate Therapeutics has received clearance from the U.S. Food and Drug Administration (FDA) to conduct two clinical trials of PROHEMA® in pediatric patients undergoing HSCT. Both the Phase 1b PROMPT study for the treatment of hematologic malignancies and the Phase 1b PROVIDE study for the treatment of inherited metabolic disorders are expected to commence enrollment in the second half of 2014.
"Over the past five years, we have worked closely with Fate Therapeutics, providing capital to support the Company's development of its hematopoietic stem cell modulation platform, and we are pleased to continue this long-standing relationship as the Company advances and expands its clinical development of PROHEMA®," said Michael White, Managing Director, Life Sciences, of Silicon Valley Bank's Southwest Division. "Our dedication to the life science and healthcare sector enables us to put financings in place that our clients, like Fate Therapeutics, need in their pursuit of therapeutics to improve patients' lives."
Proceeds from the transaction will be used for general working capital purposes including the expansion of the Company's research on therapeutic applications of human induced pluripotent stem cell (iPSC)-derived hematopoietic cells and myogenic progenitor cells. Subject to the achievement of a specified clinical milestone relating to the PUMA study, the Company has the option to access a second tranche of up to $10 million through the end of the fourth quarter of 2014. There is no warrant coverage under the first tranche of the debt facility, and 2% warrant coverage on the debt facility in the event the Company elects to access the second tranche. Assuming the full amount of the second tranche is accessed, the total cost of capital of the debt financing is approximately 11.8%, including the cost of the warrants, based on current market valuations.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the discovery and development of pharmacologic modulators of adult stem cells, including small molecules and therapeutic proteins, to treat orphan diseases. The Company has built two adult stem cell modulation platforms: a hematopoietic stem cell (HSC) modulation platform, which seeks to optimize the therapeutic potential of HSCs for treating patients with hematologic malignancies and rare genetic disorders, and a muscle satellite stem cell modulation platform, which seeks to activate the regenerative capacity of muscle for treating patients with degenerative muscle disorders. The Company is presently advancing its lead HSC product candidate, PROHEMA®, in Phase 2 clinical development for hematologic malignancies, while also advancing its proprietary Wnt7a protein analogs in preclinical development for the treatment of muscular dystrophies. Fate Therapeutics is headquartered in San Diego, CA. For more information, please visit www.fatetherapeutics.com.
Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the therapeutic potential of PROHEMA® in adult and pediatric patients with hematologic malignancies and pediatric patients with inherited metabolic disorders, our clinical development plans for PROHEMA®, our research and development of hematopoietic- and muscle-based cellular therapeutics, the availability of loan funds under our debt facility, the expected use of the loan funds received under the debt facility, and our projected cash runway. These and any other forward-looking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, the risks that the second tranche of loan funding may become unavailable to us as a result of any events of default or failure by us to satisfy certain requirements under the terms of the debt facility, PROHEMA® may not produce the therapeutic benefits suggested by the results observed in our prior preclinical and clinical development, or may cause other unanticipated adverse effects, the risk of cessation or delay of any ongoing or planned research, preclinical or clinical development activities for a variety of reasons including the uncertainty of the FDA IND review process and other regulatory requirements, additional information that may be requested or additional obligations, including changes to our clinical development plans or protocols, that may be imposed by the FDA as a condition to our initiation or continuation of clinical trials with PROHEMA®, or any adverse events or other negative results that may be observed during development, and the adequacy of available cash and available amounts under our credit facilities to meet our future liquidity needs. For a discussion of other risks and uncertainties, and other important factors, any of which could cause our actual results to differ from those contained in the forward-looking statements, see the risks and uncertainties detailed in the company's periodic filings with the Securities and Exchange Commission, including but not limited to the company's Form 10-Q for the first quarter ended March 31, 2014, and from time to time the company's other investor communications. Fate Therapeutics is providing the information in this press release as of this date and does not undertake any obligation to update any forward-looking statements contained in this press release as a result of new information, future events or otherwise.
CONTACT:
Paul Cox, Stern Investor Relations, Inc.
212.362.1200, paul@sternir.com
- Company receives multimillion dollar payment for a preclinical milestone-
Cambridge, Mass., July 30, 2014 -- Proteostasis Therapeutics, Inc., a company developing novel therapeutics that regulate protein homeostasis to improve outcomes for patients with orphan and neurodegenerative diseases, today announced that it has successfully completed a key preclinical milestone in the collaboration with Biogen Idec. Proteostasis has received a multimillion dollar milestone payment as a result of this achievement.
“We are very encouraged to reach this milestone early in our collaboration with Biogen Idec,” said Markus Haeberlein, Ph.D., Chief Scientific Officer at Proteostasis Therapeutics. “Achieving a target protein reduction through inhibition of Usp14 in a whole animal system is an important biological validation and demonstrates the disease-modifying potential of our inhibitors against a variety of neurodegenerative disorders. We look forward to continuing to work with Biogen Idec to bring this program toward candidate designation.”
Neurodegenerative diseases, such as Alzheimer’s disease and Parkinson’s disease, have elevated levels of aggregation-prone proteins in the brain. Prior research has shown that the inhibition of the deubiquinating enzyme, Usp14, enhances proteasome activity and increases the clearance of such proteins.
“We remain optimistic about Usp14 as a very interesting and potentially important target for neurodegeneration,” said Spyros Artavanis-Tsakonas, Ph.D., Chief Scientific Officer at Biogen Idec.
In December 2013, Proteostasis and Biogen Idec entered into a collaboration to research and develop therapeutic candidates based on the inhibition of Usp14. Under the agreement, the companies are conducting preclinical research to identify lead compounds for clinical development. At specified points, Proteostasis may opt in for global co-development and U.S. co-commercialization rights. The total deal contains payments of up to $200 million, as well as tiered royalties.
About Proteostasis Therapeutics
Proteostasis Therapeutics is developing disease-modifying therapeutics for cystic fibrosis and neurodegenerative diseases. The Company’s technology combines both phenotypic and target based drug discovery to develop therapeutics that modulate protein homeostasis pathways and correct for imbalances in protein folding, trafficking, and clearance. For more information, please visit www.proteostasis.com.
Investor Contacts:
Susan Shepard
Proteostasis Therapeutics, Inc.
sshepard@proteostasis.com
(617) 225-0096 x2113
Malini Chatterjee
Blueprint Life Science Group
mchatterjee@bplifescience.com
(917) 330-4269
Phase 1b PROVIDE Study in Pediatric Patients with Inherited Metabolic Disorders Expected to Begin in
4Q14
SAN DIEGO, July 29, 2014 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE), a biopharmaceutical company engaged in the discovery and development of adult stem cell modulators to treat orphan diseases, announced today that the U.S. Food and Drug Administration (FDA) has cleared its Investigational New Drug Application (IND) for the clinical development of PROHEMA® in pediatric patients undergoing hematopoietic stem cell (HSC) transplantation for the treatment of inherited metabolic disorders (IMDs). The FDA's clearance of the IND allows the Company to begin expanding its clinical investigation of PROHEMA into rare, non-malignant disorders. The Company plans to initiate enrollment of the "PROVIDE" trial (PROHEMA eValuation for the treatment of Inherited metabolic DisordErs) in pediatric patients with IMDs in the fourth quarter of 2014.
"For many severe inherited metabolic disorders, including those with central nervous system involvement, cellular enzyme replacement through unrelated donor cord blood transplantation has emerged as a potentially transformative therapeutic intervention," commented Christian Weyer, M.D., M.A.S., President and Chief Executive Officer of Fate Therapeutics. "We believe pharmacologically-optimized HSC therapeutics, such as PROHEMA, hold significant promise for improving outcomes in patients with inherited metabolic and other rare genetic disorders. FDA clearance of the PROVIDE study marks an important step towards our goal of clinically evaluating this novel treatment paradigm in the non-malignant disease setting."
The PROVIDE trial is designed to enroll up to 12 patients with various forms of IMDs, between the ages of 1 and 18, at up to three leading U.S. pediatric HSC transplant centers. The trial's inclusion criteria allows for the enrollment of patients with different types of lysosomal and peroxisomal storage diseases such as Hurler and Hunter syndromes, Krabbe disease and various other leukodystrophies, among others. The primary endpoint of the PROVIDE trial is safety as assessed by neutrophil engraftment.
PROHEMA (16, 16-dimethyl prostaglandin E2, or dmPGE2, modulated cord blood), the Company's lead pharmacologicallymodulated HSC therapeutic, is currently in Phase 2 clinical development for the treatment of adult patients with hematologic malignancies undergoing allogeneic HSC transplantation, and the Company intends to initiate enrollment of a Phase 1b clinical trial in pediatric patients with hematologic malignancy in mid-2014. In 2010, the FDA granted PROHEMA orphan designation for the enhancement of stem cell engraftment in patients undergoing allogeneic cord blood HSC transplantation. In in vivo murine models of allogeneic HSC transplantation, Fate Therapeutics demonstrated that the use of PROHEMA, as compared to unmodulated HSCs, led to a significant increase both in the engraftment of donor HSCs and in the donor-derived expression of enzyme in the brain.
About Inherited Metabolic Disorders (IMDs) and Allogeneic HSC Transplantation (HSCT)
Inherited metabolic disorders include a range of genetic enzyme deficiencies that interfere with critical metabolic pathways necessary to maintain normal organ function. In many of these disorders, the enzyme deficiency leads to cellular accumulation of toxic intermediates within the brain, causing progressive neurological damage that cannot be addressed with enzyme replacement therapy. For those inherited metabolic disorders, which include over 20 lysosomal and peroxisomal storage diseases such as Hurler and Hunter syndromes, Krabbe disease and multiple leukodystrophies, allogeneic HSCT holds potential as a one-time, definitive therapy. Following allogeneic HSCT, donor-derived cells can migrate to and engraft in the brain, providing a long-term supply of an otherwise deficient enzyme to the central nervous system in a process known as cross-correction.
About PROHEMA®
PROHEMA is a pharmacologically-modulated, cord blood-derived hematopoietic stem cell (HSC) therapeutic. PROHEMA is produced through a proprietary, two-hour, ex vivo cell modulation process that results in rapid activation of key biological pathways involved in homing, proliferation and survival of HSCs. In preclinical testing, PROHEMA has demonstrated the potential to accelerate engraftment and to drive durable hematopoietic reconstitution, without the need for multi-week expansion protocols. In an initial Phase 1b clinical trial in adult patients with hematologic malignancies undergoing double umbilical cord blood transplant (dUCBT), the median time to neutrophil recovery ( > 500 cells/μL) with PROHEMA was 17.5 days, which compares favorably to historical norms for patients undergoing dUCBT. In that trial, 100-day survival with PROHEMA was 100%, and PROHEMA provided the dominant source of hematopoiesis in 10 of 12 evaluable subjects, suggesting that treatment with PROHEMA may accelerate engraftment and drive durable and preferential hematologic reconstitution.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the discovery and development of pharmacologic modulators of adult stem cells, including small molecules and therapeutic proteins, to treat orphan diseases. The Company has built two adult stem cell modulation platforms: a hematopoietic stem cell (HSC) modulation platform, which seeks to optimize the therapeutic potential of HSCs for treating patients with hematologic malignancies and rare genetic disorders, and a muscle satellite stem cell modulation platform, which seeks to activate the regenerative capacity of muscle for treating patients with degenerative muscle disorders. The Company is presently advancing its lead HSC product candidate, PROHEMA, in Phase 2 clinical development for hematologic malignancies, while also advancing its proprietary Wnt7a protein analogs in preclinical development for the treatment of muscular dystrophies. Fate Therapeutics is headquartered in San Diego, CA. For more information, please visit www.fatetherapeutics.com.
Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the therapeutic potential of PROHEMA®, and our clinical development plans for PROHEMA®, including the timing of, and our ability to conduct, the PROVIDE and PROMPT studies. These and any other forward-looking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, the risks that the results of PROHEMA® observed in prior preclinical and clinical development may not be replicated in our PROVIDE clinical trial, or other current or subsequent clinical trials, of PROHEMA®, and that PROHEMA® may not produce the therapeutic benefits suggested by the results observed in our prior clinical development, or may cause other unanticipated adverse effects, in current or subsequent clinical trials, the risk of cessation or delay of any ongoing or planned preclinical or clinical development activities for a variety of reasons, including additional information that may be requested or additional obligations that may be imposed by the FDA as a condition to our commencement and continuation of clinical trials with PROHEMA®, any difficulties or delays in patient enrollment in the PROVIDE study, or any adverse events or other negative results that may be observed in the PROVIDE study. For a discussion of other risks and uncertainties, and other important factors, any of which could cause our actual results to differ from those contained in the forward-looking statements, see the risks and uncertainties detailed in the company's periodic filings with the Securities and Exchange Commission, including but not limited to the company's Form 10-Q
for the quarter ended March 31, 2014, and from time to time the company's other investor communications. Fate Therapeutics is providing the information in this press release as of this date and does not undertake any obligation to update any forwardlooking statements contained in this press release as a result of new information, future events or otherwise.
CONTACT:
Paul Cox, Stern Investor Relations, Inc.
212.362.1200, paul@sternir.com
SEATTLE and SOUTH SAN FRANCISCO, Calif., July 24, 2014 (GLOBE NEWSWIRE) -- Immune Design (Nasdaq:IMDZ) announced today the pricing of its initial public offering of 5,000,000 shares of its common stock at a price of $12.00 per share for an aggregate offering of $60.0 million, before underwriting discounts, commissions and expenses. All of the shares of common stock are being offered by Immune Design. In addition, Immune Design has granted the underwriters a 30-day option to purchase up to 750,000 additional shares of common stock.
The shares of Immune Design's common stock will trade on the NASDAQ Global Market under the symbol "IMDZ" beginning on July 24, 2014. The offering is expected to close on July 29, 2014, subject to customary closing conditions.
Jefferies LLC and Leerink Partners LLC are acting as joint book-running managers and Wells Fargo Securities, LLC is acting as lead manager for the offering.
A registration statement relating to the common stock being offered by Immune Design was declared effective by the Securities and Exchange Commission on July 23, 2014. The offering is being made only by means of a final prospectus, which is part of the effective registration statement. Copies of the final prospectus, when available, may be obtained by contacting Jefferies LLC, Attention: Equity Syndicate Prospectus Department, 520 Madison Avenue, 12th Floor, New York, NY 10022, telephone: (877) 547-6340, e-mail: Prospectus_Department@Jefferies.com; or Leerink Partners
LLC, Attention: Syndicate Department, One Federal Street, 37th Floor, Boston, MA 02110, by telephone at (800) 808-7525, or by email at syndicate@leerink.com.
This press release shall not constitute an offer to sell or the solicitation of an offer to buy, nor shall there be any sale of these securities in any state or jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction.
About Immune Design
Immune Design is a clinical stage immunotherapy company employing next-generation in vivo approaches to enable the body's immune system to fight disease. The company's technologies are engineered to activate the immune system's natural ability to create tumor-specific cytotoxic T cells to fight cancer and other chronic diseases. Immune Design's clinical programs are the product of its two synergistic discovery platforms: DCVexTM and GLAASTM. Immune Design has offices in Seattle, Washington and South San Francisco, California. For more information, visit www.immunedesign.com.
CONTACT:
Immune Design:
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206.769.9219
NOVATO, Calif., July 14, 2014 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (Nasdaq:RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced the closing of its public offering of 2,319,951 shares of common stock at a price to the public of $40.00 per share, which includes the exercise in full by the underwriters of their option to purchase up to 302,602 additional shares of common stock. Ultragenyx is selling 1,613,879 shares of its common stock in this offering and certain selling stockholders are selling 706,072 shares of common stock in this offering. Ultragenyx is not receiving any proceeds from the sale of any shares sold by the selling stockholders. Ultragenyx estimates net proceeds from the offering to be approximately $60.2 million, after deducting underwriting discounts and commissions and estimated offering expenses.
J.P. Morgan Securities LLC and Morgan Stanley & Co. LLC acted as the joint book-running managers for the offering. Cowen and Company, LLC acted as lead manager, and Robert W. Baird & Co. Incorporated acted as co-manager.
A registration statement relating to these securities has been filed with the Securities and Exchange Commission and was declared effective on July 8, 2014. The offering was made only by means of a written prospectus forming part of the effective registration statement. Copies of the final prospectus relating to this offering may be obtained from J.P. Morgan Securities LLC, Attention: Broadridge Financial Solutions, 1155 Long Island Avenue, Edgewood, NY 11717, or from Morgan Stanley & Co. LLC, Attention: Prospectus Department, 180 Varick Street, 2nd Floor, New York, NY 10014 or via e-mail at prospectus@morganstanley.com.
This press release shall not constitute an offer to sell or the solicitation of an offer to buy, nor shall there be any sale of these securities in any state or jurisdiction in which such offer, solicitation, or sale would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction.
About Ultragenyx
Ultragenyx is a development-stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with an initial focus on serious, debilitating genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
Forward-Looking Statements
Except for the historical information contained herein, the matters set forth in this press release are forward-looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance or achievements to differ significantly from those expressed or implied by the forward-looking statements. Such risks and uncertainties include, among others, the uncertainties related to market conditions, uncertainties inherent in the clinical drug development process, including the regulatory approval process, the timing of our regulatory filings and other matters that could affect the availability or commercial potential of our drug candidates. Ultragenyx undertakes no obligation to update or revise any forward-looking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's Registration Statement on Form S-1 filed with the Securities and Exchange Commission on June 25, 2014, as amended from time to time, and the related Prospectus, its Quarterly Report on Form 10-Q for the quarter ended March 31, 2014 filed with the Securities and Exchange Commission on May 12, 2014, and its subsequent periodic reports filed with the Securities and Exchange Commission.
CONTACT:
Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
NOVATO, Calif., July 9, 2014 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (Nasdaq:RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced the pricing of its underwritten public offering of 2,017,349 shares of its common stock at a price to the public of $40.00 per share, before underwriting discounts. Ultragenyx is selling 1,311,277 shares of its common stock in this offering, and certain selling stockholders are selling 706,072 shares of common stock in this offering. Ultragenyx will not receive any proceeds from the sale of any shares sold by the selling stockholders. In addition, the company has granted the underwriters of the offering an option for a period of 30 days to purchase up to an aggregate of an additional 302,602 shares of the company's common stock at the public offering price, less the underwriting discount.
Ultragenyx intends to use the net proceeds of the offering to continue to advance its clinical and preclinical pipeline, including potential new formulations of or indications for its existing product candidates or potential new product candidates, and to increase investment in early-stage research capabilities and general infrastructure, with any remaining proceeds to be used for other ongoing research and development, working capital and other general corporate purposes. Ultragenyx may also use a portion of the net proceeds to in-license, acquire, or invest in additional businesses, technologies, products, or assets, though Ultragenyx currently has no specific agreements, commitments, or understandings with respect to any in-licensing or acquisitions.
The offering is expected to close on or about July 14, 2014, subject to satisfaction of customary closing conditions. J.P. Morgan Securities LLC and Morgan Stanley & Co. LLC are acting as the joint book-running managers for the offering. Cowen and Company, LLC is acting as lead manager, and Robert W. Baird & Co. Incorporated is acting as co-manager.
A registration statement relating to these securities has been filed with, and declared effective by, the Securities and Exchange Commission on July 8, 2014. This offering is being made solely by means of a prospectus. Copies of the prospectus related to the offering may be obtained from J.P. Morgan Securities LLC, Attention: Broadridge Financial Solutions, 1155 Long Island Avenue, Edgewood, NY 11717, or from Morgan Stanley & Co. LLC, Attention: Prospectus Department, 180 Varick Street, 2nd Floor, New York, NY 10014.
This press release shall not constitute an offer to sell or the solicitation of an offer to buy, nor shall there be any sale of these securities in any state or jurisdiction in which such offer, solicitation, or sale would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction.
About Ultragenyx
Ultragenyx is a development-stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with an initial focus on serious, debilitating genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
Forward-Looking Statements
Except for the historical information contained herein, the matters set forth in this press release, including statements regarding the expected closing of the public offering, are forward-looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance or achievements to differ significantly from those expressed or implied by the forward-looking statements. Such risks and uncertainties include, among others, the uncertainties related to market conditions and the completion of the public offering on the anticipated terms or at all, uncertainties inherent in the clinical drug development process, including the regulatory approval process, the timing of our regulatory filings and other matters that could affect the availability or commercial potential of our drug candidates. Ultragenyx undertakes no obligation to update or revise any forward-looking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's Registration Statement on Form S-1 filed with the Securities and Exchange Commission on June 25, 2014, as amended from time to time, its Quarterly Report on Form 10-Q for the quarter ended March 31, 2014 filed with the Securities and Exchange Commission on May 12, 2014, and its subsequent periodic reports filed with the Securities and Exchange Commission.
CONTACT:
Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
NOVATO, Calif., July 1, 2014 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (Nasdaq:RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, announced the first patient screened and enrolled in the Phase 2 study of the human monoclonal anti-FGF23 antibody KRN23 (UX023) in pediatric patients with Xlinked hypophosphatemia (XLH). XLH is an inherited metabolic bone disease characterized by short stature, skeletal deformities, bone pain, fractures, and muscle weakness.
"The initiation of the Phase 2 study of KRN23 in pediatric XLH patients is a significant step forward in our overall development plan for this debilitating bone disease," commented Emil D. Kakkis, M.D., Ph.D., Ultragenyx's Chief Executive Officer. "Completed studies in adults with XLH show KRN23's effect in increasing phosphate levels and bone remodeling. Due to the high rate of growth and bone formation during childhood and the critical role phosphate plays in bone mineralization, pediatric patients with XLH may have the greatest potential for improvement from treatment with KRN23."
The multi-center, randomized, open-label, dose-finding Phase 2 clinical study will evaluate safety and efficacy in approximately 30 prepubertal pediatric XLH patients. The primary objectives of the study are to identify a dose and dosing regimen and to establish the safety profile of treatment with KRN23 in pediatric XLH patients. Preliminary clinical effects of KRN23 treatment on bone health and deformity as measured by radiographic assessments, growth, muscle strength, and motor function will also be assessed, as well as markers of bone health and patient-reported outcomes of pain, disability, and quality of life. The study has been evaluated and accepted for conduct by the United States Food and Drug Administration (FDA), the United Kingdom Medicinal and Health Regulatory Authority (MHRA), and the Dutch Medicines Evaluation Board (CBG-MEB).
The study will consist of a 16-week individual dose-titration period followed by a 48-week treatment period. The goal of the dose-titration period is to identify the individualized dose of KRN23 required to achieve stable serum phosphorus levels in the target range. Patients will be divided into three cohorts of escalating starting dose levels of KRN23 with either monthly or biweekly dosing regimens. At the end of the 16-week dose-titration period, patients will receive their individually-optimized dose of KRN23 on a monthly or biweekly basis for the 48-week treatment period. An interim analysis of safety and pharmacodynamic data will be conducted after 24 weeks of the treatment period.
In a recent meeting with Ultragenyx, the FDA agreed that blinded radiographic assessments of changes in bone abnormalities, i.e. rickets and bowing, and changes in growth may be used as primary endpoint measures in the pediatric development program and a potential Phase 3 study. The FDA also indicated that a Phase 3 study in pediatric patients could be open-label, but recommended inclusion of a standard of care control arm for comparison on a non-inferiority basis. The final design of a pediatric Phase 3 study would be determined once sufficient safety and efficacy data are available and after further consultation with the FDA. Ultragenyx also intends to continue clinical development in the adult population in parallel with the pediatric program.
About X-linked Hypophosphatemia (XLH)
XLH is a disorder of phosphate metabolism caused by phosphate wasting in the urine leading to severe hypophosphatemia. XLH is the most common heritable form of rickets that is inherited as an X-linked dominant trait affecting both males and females, though some reports indicate that the disease may be more severe in males. Studies suggest there are approximately 12,000 XLH patients in the United States. XLH is a distinctive bone disease characterized by inadequate mineralization of bone that leads to a spectrum of abnormalities, including rickets, progressive bowing of the leg, osteomalacia, bone pain, waddling gait, short stature, gross motor impairment, muscle weakness, osteopenia, frequent/poorly healing microfractures, spinal stenosis, enthesopathy, and osteoarthritis.
Most patients are managed using oral phosphate replacement and vitamin D (calcitriol) therapy, which requires frequent divided doses and careful medical monitoring. It is partially effective at reducing rickets, but it does not improve growth and can be challenging to optimize the dose without increasing the risk of depositing phosphate-calcium precipitates in the kidneys (nephrocalcinosis).
About KRN23 and FGF23
KRN23 is an investigational recombinant fully human monoclonal IgG1 antibody against the phosphaturic hormone fibroblast growth factor 23 (FGF23) being developed to treat XLH, a disease characterized by excess activity of FGF23. FGF23 is a hormone that reduces serum levels of phosphorus and vitamin D by regulating phosphate excretion and vitamin D production by the kidney. Phosphate wasting in XLH is caused by excessive levels and activity of FGF23. KRN23 is designed to bind to and thereby inhibit the excessive biological activity of FGF23. By blocking excess FGF23 in patients with XLH, KRN23 is intended to restore normal phosphate reabsorption from the kidney and increase the production of vitamin D, which enhances intestinal absorption of phosphate and calcium. Ultragenyx and Kyowa Hakko Kirin Co., Ltd. (KHK) entered into a collaboration and license agreement in August 2013 to develop and commercialize KRN23.
About Ultragenyx
Ultragenyx is a development-stage biotechnology company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with an initial focus on serious, debilitating genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
About Kyowa Hakko Kirin
Kyowa Hakko Kirin is a leading biopharmaceutical company in Japan focusing on its core business area of oncology, nephrology, and immunology/allergy. Kyowa Hakko Kirin leverages antibody-related leading-edge technologies to discover and develop innovative new drugs aiming to become a global specialty pharmaceutical company which contributes to the health and well-being of people around the world.
For more information, please visit http://www.kyowa-kirin.com.
Forward-Looking Statements
Except for the historical information contained herein, the matters set forth in this press release, including statements regarding the potential for improvement of pediatric patients with XLH as a result of treatment with KRN23, the design of the study, the number of patients who will participate in the study, the timing of completing an interim analysis of the data,expectations regarding initiation and design of a pivotal Phase 3 program, and clinical development of KRN23 in adult patients with XLH, are forward-looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance or achievements to differ significantly from those expressed or implied by the forwardlooking statements. Such risks and uncertainties include, among others, the uncertainties inherent in the clinical drug development process, including the regulatory approval process (including the possibility that the FDA could change its prior recommendation regarding the design of a Phase 3 study for KRN23 or make additional comments in further discussions with the Company), the timing of our regulatory filings and other matters that could affect the availability or commercial potential of our drug candidate. Ultragenyx undertakes no obligation to update or revise any forward-looking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's Quarterly Report on Form 10- Q for the quarter ended March 31, 2014 filed with the Securities and Exchange Commission on May 12, 2014, and its subsequent periodic reports filed with the Securities and Exchange Commission.
CONTACT:
Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
SAN DIEGO, June 30, 2014 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE), a biopharmaceutical company engaged in the discovery and development of adult stem cell modulators to treat orphan diseases, announced today that it has submitted an investigational new drug (IND) application to the U.S. Food and Drug Administration (FDA) for the clinical development of PROHEMA in pediatric patients with inherited metabolic disorders undergoing hematopoietic stem cell transplantation (HSCT). Pending FDA review of the IND application, the Company plans to initiate a Phase 1b clinical trial in pediatric patients who have inherited metabolic disorders where enzyme replacement therapy is not a viable treatment option, including certain patients with central nervous system involvement.
"Building upon the clinical development of PROHEMA in adult and pediatric patients with hematologic malignancies, we believe there are significant opportunities for development of pharmacologically-optimized HSC therapeutics in rare genetic disorders," said Christian Weyer, M.D., M.A.S., President and Chief Executive Officer of Fate Therapeutics. "Submission of this IND application to support the clinical evaluation of PROHEMA in inherited metabolic disorders reflects our commitment to improving patient outcomes across a broad spectrum of malignant and non-malignant conditions."
Inherited metabolic disorders include a range of genetic enzyme deficiencies that interfere with critical metabolic pathways necessary to maintain normal organ function. In many of these disorders, the enzyme deficiency leads to cellular accumulation of toxic intermediates within the brain, causing progressive neurological damage that cannot be addressed with enzyme replacement therapy. For those inerited metabolic disorders, which include over 20 lysosomal and peroxisomal storage diseases such as Hurler and Hunter syndromes, Krabbe disease and multiple leukodystrophies, allogeneic HSCT holds potential as a one-time, definitive therapy. Following allogeneic HSCT, donor-derived cells can migrate to and engraft in the brain, providing a long-term supply of an otherwise deficient enzyme to the central nervous system in a process known as cross-correction. In in vivo murine models of allogeneic HSCT, Fate Therapeutics has demonstrated that the use of PROHEMA, as compared to unmanipulated hematopoietic stem cells (HSCs), led to a significant increase both in the engraftment of donor HSCs and in the donor-derived expression of enzyme in the brain.
About PROHEMA
PROHEMA® (16, 16-dimethyl prostaglandin E2, or dmPGE2, modulated cord blood) is a pharmacologically-modulated, cord blood-derived hematopoietic stem cell (HSC) therapeutic. PROHEMA is produced through a proprietary, two-hour, ex vivo cell modulation process that results in rapid activation of key biological pathways involved in homing, proliferation and survival of HSCs. In preclinical testing, PROHEMA has demonstrated the potential to accelerate engraftment and to drive durable hematopoietic reconstitution, without the need for multi-week expansion protocols. In an initial Phase 1b clinical trial in adult patients with hematologic malignancies undergoing double umbilical cord blood transplant (dUCBT), the median time to neutrophil recovery ( > 500 cells/μL) with PROHEMA was 17.5 days, which compares favorably to historical norms for patients undergoing dUCBT. In that trial, 100-day survival with PROHEMA was 100%, and PROHEMA provided the dominant source of hematopoiesis in 10 of 12 evaluable subjects, suggesting that treatment with PROHEMA may accelerate engraftment and drive durable and preferential hematologic reconstitution.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the discovery and development of pharmacologic modulators of adult stem cells, including small molecules and therapeutic proteins, to treat orphan diseases. The Company has built two adult stem cell modulation platforms: a hematopoietic stem cell (HSC) modulation platform, which seeks to optimize the therapeutic potential of HSCs for treating patients with hematologic malignancies and rare genetic disorders, and a muscle satellite stem cell modulation platform, which seeks to activate the regenerative capacity of muscle for treating patients with degenerative muscle disorders. The Company is presently advancing its lead HSC product candidate, PROHEMA, in Phase 2 clinical development for hematologic malignancies, while also advancing its proprietary Wnt7a protein analogs in preclinical development for the treatment of muscular dystrophies. Fate Therapeutics is headquartered in San Diego, CA. For more information, please visit www.fatetherapeutics.com.
Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the therapeutic potential of PROHEMA, including in pediatric patients who have inherited metabolic disorders, and our clinical development plans for PROHEMA, including our ability to initiate and conduct our planned Phase 1b clinical trial in this patient population. These and any other forward-looking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, the risks that the results of PROHEMA observed in prior preclinical and clinical development may not be replicated in other current or subsequent clinical trials of PROHEMA, and that PROHEMA may not produce the therapeutic benefits suggested by the results observed in our prior preclinical and clinical development, or may cause other unanticipated adverse effects, in current or subsequent clinical trials, the risk of cessation or delay of any ongoing or planned preclinical or clinical development activities for a variety of reasons, including the uncertainty of the FDA IND review process and other regulatory requirements, additional information that may be requested or additional obligations, including changes to our clinical development plans or protocols, that may be imposed by the FDA as a condition to our initiation or continuation of clinical trials with PROHEMA, any difficulties or delays in patient enrollment in clinical trials, or any adverse events or other negative results that may be observed in clinical trials. For a discussion of other risks and uncertainties, and other important factors, any of which could cause our actual results to differ from those contained in the forward-looking statements, see the risks and uncertainties detailed in the company's periodic filings with the Securities and Exchange Commission, including but not limited to the company's Form 10-Q for the first quarter ended March 31, 2014, and from time to time the company's other investor communications. Fate Therapeutics is providing the information in this press release as of this date and does not undertake any obligation to update any forward-looking statements contained in this press release as a result of new information, future events or otherwise.
CONTACT:
Paul Cox, Stern Investor Relations, Inc.
212.362.1200, paul@sternir.com
Source:
Fate Therapeutics, Inc
News Provided by Acquire Media
Treatment With KRN23 Induces a Sustained Increase in Serum Phosphorus and Increases in Bone Remodeling Markers
NOVATO, Calif., June 24, 2014 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (Nasdaq:RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced the presentation of results from a multiple-dose study, conducted by Kyowa Hakko Kirin Pharma, Inc. (KKP), of the investigational anti-FGF23 monoclonal antibody KRN23 (UX023) in adult patients with X-linked hypophosphatemia (XLH). XLH is an inherited metabolic bone disease characterized by short stature, skeletal deformities, bone pain, fractures, and muscle weakness. The data was presented at the 2014 ICE/ENDO joint meeting of The Endocrine Society and The International Congress of Endocrinology in Chicago.
"We are pleased with the results of the Phase 1/2 study of KRN23 in adult patients with XLH," commented Emil D. Kakkis, M.D., Ph.D., Ultragenyx's Chief Executive Officer. "Based on observed improvements in phosphate metabolism, bone remodeling markers, and certain quality of life measures, we are encouraged by the potential for KRN23 to treat the underlying cause of bone disease in both adult and pediatric patients with XLH."
A multiple-dose Phase 1/2 study (INT-001) with up to four escalating doses from 0.05 mg/kg to 0.6 mg/kg was completed in 28 adult XLH patients. The study assessed the safety and tolerability of subcutaneous injections of KRN23 given once every four weeks.
The efficacy data from the Phase 1/2 study demonstrate that blocking excess FGF23 increases urinary phosphorus
reabsorption, serum phosphorus levels, and 1,25 dihydroxy vitamin D levels. Repeat doses with KRN23 over four months led to an increase in serum phosphorus in 100% of patients, with approximately 89% of patients reaching the low end of the normal range. Peak mean serum phosphorus increased to 3.03 ± 0.42 mg/dL after the fourth dose, which is an approximately 60% increase from the mean of 1.89 ± 0.33 mg/dL at baseline. Comparable increases were observed in mean reabsorption of phosphate from the urine (TmP/GFR) and mean serum 1,25 dihydroxy vitamin D levels.
Increases in bone remodeling markers were also observed, including markers of bone formation and bone resorption. The increase in P1NP (procollagen type I N propeptide) from baseline was statistically significant (p < 0.05) after all doses and the increase in osteocalcin was statistically significant (p < 0.05) after the fourth dose. Increases in BALP (bone alkaline phosphatase) and CTx (carboxy terminal cross-linked telopeptide of type I collagen) were also observed. These data support the concept that KRN23's impact on improving phosphate metabolism will improve bone remodeling, a critical part of creating strong, properly-formed bones.
Patients completed two quality of life questionnaires at baseline and after the fourth dose; mean scores improved for all SF- 36v21 and WOMAC2 scales, consistent with an improvement in health status for both instruments, although these were not blinded assessments. For SF-36v2, statistically significant increases from baseline were observed in the role limitations due to physical health, bodily pain, and physical component summary scales. For WOMAC, statistically significant increases from baseline were observed in the physical functioning and stiffness scales. Stiffness is one of the major symptoms of XLH in adult patients. These findings will be evaluated objectively in a future randomized controlled study.
There were no significant changes in parathyroid hormone, serum calcium, or urinary calcium excretion, consistent with the Phase 1 data showing that KRN23 can specifically improve phosphate control without interfering with calcium control. The most common adverse events were nasopharyngitis, joint pain, diarrhea, back pain, and restless leg syndrome. Joint pain and back pain are both known symptoms of XLH in adults. There were no serious adverse events related to treatment or renal or cardiac tissue calcification. One patient discontinued treatment due to injection site reaction. No anti-KRN23 antibodies were observed.
In addition to the four-dose Phase 1/2 study (INT-001) presented at ICE/ENDO, a long-term Phase 1/2 study (INT-002) was completed to evaluate KRN23 treatment for an additional 12 doses in 22 of the patients from INT-001. Administration of KRN23 continued to show a favorable safety profile and sustained improvements in phosphate metabolism and other measures. Data from the long-term Phase 1/2 study is expected to be presented at the American Society for Bone and Mineral Research (ASBMR) Annual Meeting in September.
Based on the Phase 1 and Phase 1/2 study results to date, Ultragenyx and Kyowa Hakko Kirin Co., Ltd. (KHK) plan to initiate a Phase 2 study of KRN23 in pediatric patients in 2014 and expect to continue the clinical development of KRN23 in adults with XLH.
1Medical Outcomes Study 36-item Short Form, Version 2 (SF-36v2) is comprised of eight scales measuring physical function, role limitations due to physical health, bodily pain, general health perceptions, vitality, role limitations due to emotional problems, and mental health, as well as physical component summary and mental component summary composite scores.
2Western Ontario and McMaster University Osteoarthritics Index (WOMAC) is comprised of three scales measuring pain, stiffness, and physical functioning.
About X-linked Hypophosphatemia (XLH)
XLH is a disorder of phosphate metabolism caused by phosphate wasting in the urine leading to severe hypophosphatemia. XLH is the most common heritable form of rickets that is inherited as an X-linked dominant trait affecting both males and females, though some reports indicate that the disease may be more severe in males. Studies suggest there are approximately 12,000 XLH patients in the United States. XLH is a distinctive bone disease characterized by inadequate mineralization of bone that leads to a spectrum of abnormalities, including rickets, progressive bowing of the leg, osteomalacia, bone pain, waddling gait, short stature, gross motor impairment, muscle weakness, osteopenia, frequent/poorly healing microfractures, spinal stenosis, enthesopathy, and osteoarthritis.
Most patients are managed using oral phosphate replacement and vitamin D (calcitriol) therapy, which requires frequent divided doses and careful medical monitoring. It is partially effective at reducing rickets, but it does not improve growth and can be challenging to optimize the dose without increasing the risk of depositing phosphate-calcium precipitates in the kidneys (nephrocalcinosis).
About KRN23 and FGF23
RN23 is an investigational recombinant fully human monoclonal IgG1 antibody discovered by KHK against the phosphaturic hormone fibroblast growth factor 23 (FGF23) being developed to treat XLH, a disease characterized by excess activity of FGF23. FGF23 is a hormone that reduces serum levels of phosphorus and vitamin D by regulating phosphate excretion and vitamin D production by the kidney. Phosphate wasting in XLH is caused by excessive levels and activity of FGF23. KRN23 is designed to bind to and thereby inhibit the excessive biological activity of FGF23. By blocking excess FGF23 in patients with XLH, KRN23 is intended to restore normal phosphate reabsorption from the kidney and increase the production of vitamin D, which enhances intestinal absorption of phosphate and calcium. Ultragenyx and KHK entered into a collaboration and license agreement in August 2013 to develop and commercialize KRN23.
About Ultragenyx
Ultragenyx is a development-stage biotechnology company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with an initial focus on serious, debilitating genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
About Kyowa Hakko Kirin
Kyowa Hakko Kirin is a leading biopharmaceutical company in Japan focusing on its core business area of oncology, nephrology, and immunology/allergy. Kyowa Hakko Kirin leverages antibody-related leading-edge technologies to discover and develop innovative new drugs aiming to become a global specialty pharmaceutical company which contributes to the health and well-being of people around the world.
For more information, please visit http://www.kyowa-kirin.com.
Forward-Looking Statements
Except for the historical information contained herein, the matters set forth in this press release, including statements regarding the potential for KRN23 to treat the underlying cause of bone disease in adult and pediatric patients with XLH, the effects of blocking excess FGF23, the effects of improving phosphate metabolism, the evaluation of findings in future studies, the timing of release of additional data, plans to conduct additional studies (and anticipated timing of the initiation of such studies), the number of patients in the United States who have XLH and the intended result of administration of KRN23, are forward-looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance, or achievements to differ significantly from those expressed or implied by the forward-looking statements. Such risks and uncertainties include, among others, the uncertainties inherent in the clinical drug development process, including the regulatory approval process, the timing of our regulatory filings, that these results (which were in adult patients) may not translate into similar safety or efficacy in pediatric patients, and other matters that could affect the availability or commercial potential of our drug candidates. Ultragenyx undertakes no obligation to update or revise any forward-looking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's Quarterly Report on Form 10-Q filed with the Securities and Exchange Commission on May 12, 2014, and its subsequent periodic reports filed with the Securities and Exchange Commission.
CONTACT:
Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
SAN DIEGO, June 5, 2014 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE), a biopharmaceutical company engaged in the discovery and development of adult stem cell modulators to treat orphan diseases, today announced that potential clinical applications of its ex vivo approach to pharmacologically modulating hematopoietic cells will be presented at the 12th Annual International Cord Blood Symposium being held from June 5-7, 2014 in San Francisco, CA.
Two presentations will summarize the potential clinical benefits of modulating hematopoietic stem cells (HSCs) and T cells ex vivo, prior to their administration to patients undergoing allogeneic HSC transplantation (HSCT), using 16,16-dimethyl prostaglandin E2 (dmPGE2), a small molecule derivative of the naturally secreted homeostatic factor prostaglandin E2. In addition, a poster presentation will highlight the potential of the Company's proprietary small molecule-based platform to generate clinical-grade, human induced pluripotent stem cell (hiPSC)-derived therapeutics from an attached segment of an umbilical cord blood unit, while leaving the cord blood unit itself intact and available for later use in HSCT. Fate Therapeutics is currently advancing PROHEMA®, a pharmacologically-modulated HSC therapeutic, in a randomized, controlled Phase 2 clinical trial in adult patients undergoing HSCT for the treatment of hematologic malignancies.
12th Annual International Cord Blood Symposium Presentations
- Future Horizons for the Ex Vivo Modulation of Cord Blood. On Thursday, June 5th, Dan Shoemaker, Ph.D., Chief Technology Officer of Fate Therapeutics, will give a presentation on "Future Horizons for the Ex Vivo Modulation of Cord Blood" as part of a session entitled "Derivative Products from Cord Blood and Placental Tissue" to be held from 1:30 - 3:30 pm. This presentation will explore current and future clinical applications of pharmacologically-modulated umbilical cord blood, including using the Company's therapeutic candidates, in patients with hematologic malignancies and rare genetic disorders.
- PGE2-Modulated Umbilical Cord Blood Transplantation. On Saturday, June 7th, Corey Cutler, MD, MPH, FRCPC of the Dana Farber Cancer Institute (DFCI) will give a presentation from 11:00 - 11:20 am entitled "PGE2-Modulated Umbilical Cord Blood Transplantation", which will include a review of the enhanced biological properties and potential clinical benefits of pharmacologically-modulated HSCs and T cells, including PROHEMA®. Dr. Cutler served as the principal investigator for the Company's Phase 1b clinical trial of PROHEMA® in adult patients with hematologic malignancies undergoing double umbilical cord blood transplant, which trial demonstrated safety and provided encouraging indications of efficacy including an acceleration in the median time to neutrophil engraftment, an increase in the rate of overall survival at 100-days, and a decrease in the rate of cytomegalovirus reactivation, as compared to historical rates observed at the DFCI and reported in the literature.
- hiPSC Generation from Umbilical Cord Blood Segments. On Thursday, June 5th, Bahram Valamehr, Ph.D, Associate Director of Reprogramming Biology of Fate Therapeutics, will present a poster from 5:30 - 6:30 pm entitled "hiPSC Generation from Individual Umbilical Cord Blood Sample Segments for Allogeneic cGMP Banking and Cell Therapy Applications". The poster will highlight the use of the Company's proprietary small molecule-based platform to generate transgene-free, genomically-stable hiPSCs from a single frozen segment of a banked umbilical cord blood unit. These findings demonstrate the potential of using attached segments from banked umbilical cord blood units to derive, maintain and bank clinical-grade hiPSC lines without compromising the unit itself.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the discovery and development of pharmacologic modulators of adult stem cells to treat orphan diseases. The Company utilizes established pharmacologic modalities, including small molecules and therapeutic proteins, and well-characterized biological mechanisms to enhance the therapeutic potential of adult stem cells. The Company has built two adult stem cell modulation platforms: a hematopoietic stem cell (HSC) modulation platform, which seeks to optimize the therapeutic potential of HSCs for treating patients with hematologic malignancies and rare genetic disorders, and a muscle satellite stem cell modulation platform, which seeks to activate the regenerative capacity of muscle for treating patients with degenerative muscle disorders. The Company is presently advancing its lead product candidate, PROHEMA ®, a pharmacologically-modulated HSC therapeutic, in Phase 2 clinical development for hematologic malignancies. Fate Therapeutics is also advancing its proprietary Wnt7a protein analogs in preclinical development for the treatment of muscular dystrophies. Fate Therapeutics is headquartered in San Diego, CA. For more information, please visit www.fatetherapeutics.com.
Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the potential mechanisms and therapeutic benefits and applications of our programs for the modulation of adult stem cells and other cell types, our preclinical and clinical development plans including the development of PROHEMA® for the treatment of hematologic malignancies and rare genetic disorders and the development of hiPSC-based therapeutics, and the potential therapeutic benefits of pharmacological modulation of HSCs and T cells. These and any other forward-looking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, the risks that the results observed in prior clinical development may not be replicated in current or subsequent clinical trials of PROHEMA®, that PROHEMA® may not produce the therapeutic benefits suggested by the results observed in preclinical investigation or our prior clinical development, or may cause other unanticipated adverse effects in subsequent clinical trials, and of cessation or delay of any ongoing or planned preclinical or clinical development activities for a variety of reasons, including additional information that may be requested or additional obligations that may be imposed by the FDA as a condition to our initiation of new clinical trials or continuation of clinical trials with PROHEMA®, or any delays in enrollment of or negative results in clinical trials with PROHEMA®. For a discussion of other risks and uncertainties, and other important factors, any of which could cause our actual results to differ from those contained in the forward-looking statements, see the risks and uncertainties detailed in the company's periodic filings with the Securities and Exchange Commission, including but not limited to the company's Form 10-Q for the first quarter ended March 31, 2014, and from time to time the company's other investor communications. Fate Therapeutics is providing the information in this press release as of this date and does not undertake any obligation to update any forward-looking statements contained in this press release as a result of new information, future events or otherwise.
CONTACT:
Paul Cox, Stern Investor Relations, Inc.
212.362.1200, paul@sternir.com
In vivo T cell active immunotherapy generated from DCVexTM platform
June 5, 2014, Seattle, WA and South San Francisco, CA – Immune Design, a clinical-stage immunotherapy company focused on the development of novel immune-based therapies for cancer and other chronic conditions, today announced treatment of the first patient in a Phase 1 clinical trial of LV305, an immuno-oncology investigational agent from the company’s DCVexTM lentiviral vector platform.
The Phase 1 open label, multi-center trial (NCT02122861) is designed to evaluate the safety, tolerability and immunogenicity of LV305 in patients with locally advanced, relapsed, or metastatic breast cancer, melanoma, non-small cell lung cancer, ovarian cancer or sarcoma. The trial will enroll up to 36 patients at several clinical centers in the United States.
“The advancement of novel immuno-oncology agents such as LV305 that induce a tumorspecific in vivo T-cell response holds promise for the development of new and targeted approaches to cancer treatment,” said Seth M. Pollack, M.D., Principal Investigator at the Fred Hutchinson Cancer Research Center.
“LV305, which targets the NY-ESO-1 antigen expressed in a number of tumors, offers a targeted, tumor-specific in vivo approach to activating a T-cell immune response that we believe may provide benefit to a wide range of cancer patients,” said Carlos Paya, M.D., Ph.D., President and Chief Executive Officer at Immune Design. “LV305 is an integral part of our prime-boost strategy that is designed to provide a superior approach to fighting cancer. Data from the trial will include immunogenicity and initial indications of efficacy, and is intended to support the combination of LV305 with a second proprietary agent, G305, into our prime-boost strategy known as CMB305. We intend to commence a Phase 1 trial for CMB by the end of 2014.”
“I am excited to see that a novel and perhaps bold idea has matured into a product candidate now being clinically evaluated, and believe that using a novel vector of this type to deliver genetic information specifically to dendritic cells opens a new avenue that holds much promise for treating cancer,” said David Baltimore, Ph.D., President Emeritus, Robert Andrews Millikan Professor of Biology, California Institute of Technology.
About LV305
LV305, generated from Immune Design’s DCVex platform, is designed to activate the immune system through the in vivo generation of cytotoxic T cells (CTLs) initially against a specific tumor-associated antigen, NY-ESO-1. DCVex originated from underlying discoveries made by one of Immune Design’s founders and Nobel laureate, David Baltimore, Ph.D. Dr. Baltimore and his colleagues theorized that a lentiviral vector could be engineered to selectively deliver specific genetic information of a tumor antigen directly to dendritic cells to mount an immune response. LV305 is part of Immune Design’s “Specific Antigen” approach, which drives the in vivo generation of a strong, antigen-specific CTL response against selected antigens present in a tumor. Preclinical tests have demonstrated the ability of LV305 to reduce tumor growth of NYESO- 1-expressing tumors, increase production of antigen-specific CD8 cells, and significantly improve the survival of tumor-bearing animals. LV305 is the first step in Immune Design’s novel prime-boost approach to immuno-oncology, which includes combination with G305, generated from the GLAASTM platform, to expand CTLs and potentially generate a potent, durable immune response.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body’s immune system to fight disease. The company’s technologies are engineered to activate the immune system’s natural ability to create tumor-specific cytotoxic T cells to fight cancer and other chronic diseases. Immune Design’s clinical programs are the product of its two synergistic discovery platforms: DCVexTMand GLAASTM. Immune Design has offices in Seattle, Washington and South San Francisco, California. For more information, visit www.immunedesign.com.
Cautionary Note on Forward-Looking Statements
This press release contains forward looking statements. Forward looking statements contained in this press release include, but are not limited to, statements about the timing and results of the Phase 1 trial of LV305, patient enrollment of that trial and the timing of commencement of clinical trails of CMB305. Words such as "may," "will," "expect," "plan," "anticipate," "estimate," "intend" and similar expressions (as well as other words or expressions referencing future events, conditions or circumstances) are intended to identify forward looking statements. These forward looking statements are not guarantees of future performance and involve a number of unknown risks, assumptions, uncertainties and factors that are beyond Immune Design’s control. All forward looking statements are based on Immune Design’s expectations and assumptions as of the date of this press release. Actual results may differ materially from these forward looking statements. Except as required by law, Immune Design expressly disclaims any responsibility to update any forward looking statement contained herein, whether as a result of new information, future events or otherwise.
# # #
Contact:
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206.769.9219
May 27, 2014, Seattle, WA and San Francisco, CA – Immune Design, a clinical-stage immunotherapy company focused on the development of novel immune-based therapies for cancer and other chronic conditions, today announced the application of its GLAASTM discovery platform in the Phase 1 clinical trial of MEDI7510, an investigational agent for respiratory syncytial virus (RSV) being developed by MedImmune, the global biologics research and development arm of AstraZeneca. MEDI7510 is composed of MedImmune’s RSV sF antigen plus GLA, a synthetic molecule licensed from Immune Design’s GLAAS discovery platform.
This follows an existing agreement between the two companies, in which Immune Design granted MedImmune an exclusive license to use GLA to develop and commercialize vaccines in three different infectious disease indications, one of which is RSV. The parties have not disclosed the other two indications.
The Phase 1a, double-blind, randomized, placebo-controlled cohort escalation study (NCT02115815) is designed to evaluate the safety, tolerability and immunogenicity of a single ascending intramuscular dose of MEDI7510 or placebo in adults 60 years or older who are healthy or who have stable, underlying medical conditions. The trial is expected to enroll 144 patients at several clinical centers in the United States.
“We are pleased to see another potential therapy progressing in the clinic that includes GLA,” said Carlos Paya, M.D., Ph.D., President and Chief Executive Officer at Immune Design. “While we are pursuing product candidates from GLAAS in immuno-oncology, the platform also holds promise in infectious, allergic and autoimmune diseases, and we plan to continue to explore additional collaborations outside of oncology.”
About GLAAS
Immune Design’s GLAAS platform works in vivo and is based on a small synthetic molecule called GLA, which stands for glucopyranosyl lipid adjuvant. GLA selectively binds to the TLR4 receptor and causes potent activation of dendritic cells. When GLA is accompanied by an antigen and injected into a patient, the combination is taken up by DCs and leads to the production and expansion of immune cells called CD4 T helper lymphocytes. These CD4 cells play a key role in boosting the anti-tumor immune response by expanding the number and function of existing CTLs that are specific to the same antigen; and providing help to other immune cells, including B lymphocytes that are the precursor to antibodies, and natural killer cells that are also important in the overall immune response. Immune Design believes that GLAAS product candidates have the potential to target multiple types of cancer, as well as infectious, allergic and autoimmune diseases.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing next-generation in vivo approaches to enable the body’s immune system to fight disease. The company’s technologies are engineered to activate the immune system’s natural ability to create tumor-specific cytotoxic T cells to fight cancer and other chronic diseases. Immune Design’s clinical programs are the product of its two synergistic discovery platforms: DCVexTMand GLAASTM. Immune Design has offices in Seattle, Washington and South San Francisco, California. For more information, visit www.immunedesign.com.
# # #
Contact for Immune Design:
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206.769.9219

DUBLIN and SAN DIEGO, May 12, 2014 /PRNewswire/ --
- Acquisition of Lumena Pharmaceuticals, a biopharmaceutical company with late stage rare disease pipeline assets
- Adds to Shire's rare diseases portfolio and leverages this expertise, and is a perfect combination with Shire's already strong Gastrointestinal (GI) presence
- Adds LUM001 in Phase 2 development for four rare and devastating hepatic diseases, two pediatric and two adult with a potential 2016 approval and LUM002 a Phase 2-ready candidate for the treatment of non-alcoholic steatohepatitis (NASH)
- Very attractive opportunity to develop treatments for significant unmet need in rare cholestatic liver diseases as well as a treatment for non-alcoholic steatohepatitis (NASH)
- Shire will acquire Lumena Pharmaceuticals for an upfront payment of $260 million in cash, plus a payment for net cash at closing, and near-term contingent milestone payments related to ongoing clinical trials
- These two compounds, and Shire's full portfolio, will be discussed in more detail at an Investor Day later in 2014.
Shire plc (LSE: SHP,NASDAQ: SHPG) and Lumena Pharmaceuticals, Inc., a biopharmaceutical company with rare disease pipeline assets, announce the acquisition of Lumena Pharmaceuticals by Shire.
Chief Executive of Shire, Flemming Ornskov, MD comments:
"Our pipeline and strategic focus on rare diseases is even further strengthened with the acquisition of Lumena Pharmaceuticals, which also complements our strong GI presence. These attractive potential treatments may offer new hope to patients with rare cholestatic liver disease and further contribute to Shire's future growth. We are excited by the possibilities of these new assets in liver disease. We have the resources, the infrastructure and the operating capacity to invest in these new potential growth drivers which add further value to Shire's innovative pipeline."
President and CEO of Lumena Pharmaceuticals, Mike Grey comments:
"I believe that this transaction is a significant win for all parties involved, especially the patients, and the future of LUM001 as a treatment for rare cholestatic liver diseases looks brighter than ever. Shire has deep rare disease experience, a global infrastructure, and the commercial expertise to deliver LUM001 to patients around the world. The Lumena team will work closely with Shire to finish the ongoing Phase 2 clinical programs as part of our commitment to the patient populations we have championed since we formed Lumena Pharmaceuticals."
Strategic rationale and background on Lumena Pharmaceuticals
The acquisition of Lumena strengthens Shire's already valuable and robust pipeline. It complements Shire's strategic focus on Rare Diseases and provides a future growth path for Shire's Gastrointestinal business, which generated revenues of over $800 million in 2013. In acquiring Lumena, Shire is gaining experience in liver disease with the opportunity to leverage its existing GI commercial infrastructure. In addition, there is a good fit with Shire's recent acquisition of Fibrotech, which has brought pipeline programs to address unmet patient need in other fibrotic conditions including renal impairment.
Lumena Pharmaceuticals brings to Shire two new oral therapeutic compounds; LUM001, in Phase 2 with four potential orphan indications and LUM002, ready to enter Phase 2 later in 2014.
LUM001 and LUM002 are both inhibitors of the apical sodium-dependent bile acid transporter (ASBT), which is primarily responsible for recycling bile acids from the intestine to the liver. Blocking bile acid transport with ASBT inhibitors reduces bile acid absorption and has the potential to improve liver function and relieve disease symptoms (such as extreme itching associated with cholestatic liver diseases), and may slow disease progression.
These rare cholestatic liver diseases are primarily treated by hepatologists and gastroenterologists and can be covered by a small, specialty sales force consistent with Shire's model.
Shire does not expect the acquisition of Lumena to result in a change to its previously stated earnings guidance for 2014.
About LUM001
LUM001 is a novel, once-daily, orally-administered, potent and selective ASBT inhibitor that works by preventing recycling of bile acids back to the liver and is thought to reduce bile acid accumulation, improve liver function and potentially relieve the extreme itching associated with cholestatic liver disease.
LUM001 is currently in Phase 2 clinical development for four rare cholestatic liver disease indications; two pediatric and two adult with a potential 2016 launch. These potential indications are: Alagille syndrome (ALGS), progressive familial intrahepatic cholestasis (PFIC), primary biliary cirrhosis (PBC) and primary sclerosing cholangitis (PSC).
Some of the key characteristics of cholestatic liver diseases are elevated bile acids, leading to progressive liver damage that can cause liver failure, and pruritus, or severe itching. Pruritus is generally the most debilitating symptom afflicting children and adults with these diseases.
Surgical intervention, which lowers bile acid levels, has been shown to relieve symptoms and slow disease progression in patients with ALGS and PFIC - this is currently the only treatment option for these patients. Patients with cholestatic liver diseases may ultimately require liver transplants.
By reducing serum bile acids, LUM001 may offer a novel therapeutic approach for alleviating the pruritus and progressive liver damage associated with cholestatic liver diseases.
The prevalence of each of the four diseases is as follows:
| Indication US | Indication US prevalence 3 per 100,000 |
EU prevalence |
| ALGS | 9476 individuals <1 per 100,000 |
3 per 100,000 |
| PFIC | 3159 individuals 40 per 100,000 |
1 per 100,000 |
| PBC | 126,979 individuals 14 per 100,000 |
30 per 100,000 |
| PSC | 44,222 individuals 3 per 100,000 |
3 per 100,000 |
(Source: Lumena Pharmaceuticals Applications for Orphan Designation):
LUM001 has received orphan drug designation for all four potential indications in both the United States and the European Union.
About LUM002
LUM002 is a novel, once daily, orally-administered, highly potent and selective inhibitor of ASBT, in development for the treatment of nonalcoholic steatohepatitis (NASH), a common and often "silent" liver disease characterized by fat deposits in the liver and inflammation which can progress to significant fibrosis.
While the underlying cause of liver injury in NASH is not fully known, it is strongly associated with obesity, Type 2 diabetes, high cholesterol and triglycerides, and other metabolic disorders. Approximately 6 million individuals in the US are estimated to have progressed to NASH and some 600,000 to NASH-related cirrhosis (Source: World Gastroenterology Organisation Global Guidelines June 2012).
By blocking bile acid reabsorption, LUM002 is thought to modulate colonic bile acid concentrations and receptor signaling on cells in the lower portion of the GI tract. This signaling is believed to result in the secretion of peptides that regulate insulin release from the pancreas, glucose metabolism and the synthesis of cholesterol and fatty acids.
Therapeutic strategies aimed at modulating insulin resistance and normalizing lipoprotein metabolism have significant potential to benefit patients with NASH.
Phase 1 safety trials in healthy volunteers and a Phase 1b trial in patients with metabolic disease have been completed. The next step is to initiate a Phase 2 clinical trial in patients with NASH - anticipated to begin in the second half of 2014.
NOTES TO EDITORS
About Lumena Pharmaceuticals
Lumena Pharmaceuticals is a privately held, San Diego-based, biopharmaceutical company founded in 2011 by Pappas Ventures. Other investors include Alta Partners, RiverVest Venture Partners, New Enterprise Associates, Adage Capital Management and RA Capital Management.
Citi acted as the exclusive financial advisor to Lumena.
About Shire plc
Shire enables people with life-altering conditions to lead better lives.
Our strategy is to focus on developing and marketing innovative specialty medicines to meet significant unmet patient needs.
We provide treatments in Neuroscience, Rare Diseases, Gastrointestinal and Internal Medicine and we are developing treatments for symptomatic conditions treated by specialist physicians in other targeted therapeutic areas.
FORWARD - LOOKING STATEMENTS - "SAFE HARBOR" STATEMENT UNDER THE PRIVATE SECURITIES LITIGATION REFORM ACT OF 1995
Statements included in this announcement that are not historical facts are forward-looking statements. Forward-looking statements involve a number of risks and uncertainties and are subject to change at any time. In the event such risks or uncertainties materialize, Shire's results could be materially adversely affected. The risks and uncertainties include, but are not limited to, that:
- Shire's products may not be a commercial success;
- revenues from ADDERALL XR are subject to generic erosion and revenues from INTUNIV will become subject to generic competition starting in December 2014;
- the failure to obtain and maintain reimbursement, or an adequate level of reimbursement, by third-party payors in a timely manner for Shire's products may impact future revenues, financial condition and results of operations;
- Shire conducts its own manufacturing operations for certain of its Rare Diseases products and is reliant on third party contractors to manufacture other products and to provide goods and services. Some of Shire's products or ingredients are only available from a single approved source for manufacture. Any disruption to the supply chain for any of Shire's products may result in Shire being unable to continue marketing or developing a product or may result in Shire being unable to do so on a commercially viable basis for some period of time.
- the development, approval and manufacturing of Shire's products is subject to extensive oversight by various regulatory agencies and regulatory approvals or interventions associated with changes to manufacturing sites, ingredients or manufacturing processes could lead to significant delays, increase in operating costs, lost product sales, an interruption of research activities or the delay of new product launches;
- the actions of certain customers could affect Shire's ability to sell or market products profitably. Fluctuations in buying or distribution patterns by such customers can adversely impact Shire's revenues, financial conditions or results of operations;
- investigations or enforcement action by regulatory authorities or law enforcement agencies relating to Shire's activities in the highly regulated markets in which it operates may result in the distraction of senior management, significant legal costs and the payment of substantial compensation or fines;
- adverse outcomes in legal matters and other disputes, including Shire's ability to enforce and defend patents and other intellectual property rights required for its business, could have a material adverse effect on Shire's revenues, financial condition or results of operations;
- Shire faces intense competition for highly qualified personnel from other companies, academic institutions, government entities and other organizations. Shire is undergoing a corporate reorganization and the consequent uncertainty could adversely impact Shire's ability to attract and/or retain the highly skilled personnel needed for Shire to meet its strategic objectives;
- failure to achieve Shire's strategic objectives with respect to the acquisition of ViroPharma Incorporated may adversely affect Shire's financial condition and results of operations; and other risks and uncertainties detailed from time to time in Shire's filings with the U.S. Securities and Exchange Commission, including its most recent Annual Report on Form 10-K.
For further information please contact:
Shire
Investor Relations:
Jeff Poulton
jpoulton@shire.com
+1-781-482 0945
Sarah Elton-Farr
seltonfarr@shire.com
+44-1256-894157
Media:
Jessica Mann
jmann@shire.com
+44-1256-894-280
Gwen Fisher
gfisher@shire.com
+1-484-595-9836
Lumena Media contact:
Heidi Chokeir, Ph.D.
Canale Communications
Heidi@canalecomm.com
+1-619-203-5391
SOURCE Shire plc and Lumena Pharmaceuticals
Link between lysosome-based genetic disorders and neurodegenerative diseases to be explored as foundation for new therapeutics
CAMBRIDGE, Mass. — May 12, 2014 — Lysosomal Therapeutics Inc. (LTI), a company leveraging its expertise in lysosomal biology to develop novel small molecules for use in the treatment of neurodegenerative diseases, announced today it has raised $4.8 million in seed funding. Atlas Venture was the lead investor, with additional participation from Hatteras Venture Partners, Lilly Ventures, Sanofi-Genzyme BioVentures, Roche Venture Fund, Partners Innovation Fund and several angel investors, including Orion Equity Partners, LLC. LTI was founded by Dimitri Krainc, M.D., Ph.D., and former Genzyme executives Henri Termeer, Bob Carpenter and Peter Wirth. Kees Been is the founding president and chief executive officer of the company.
LTI’s unique approach to discovering new drugs for neurodegenerative diseases is based on research performed in Krainc’s lab at Massachusetts General Hospital (MGH), a founding member of Partners HealthCare and an affiliate of Harvard Medical School. The work completed at MGH by Krainc and Joseph Mazzulli, Ph.D., LTI’s scientific co-founder and a former postdoctoral fellow in the lab, builds on the association between Gaucher disease (GD), a rare lysosomal storage disorder (LSD) caused by mutations in the gene for glucocerebrosidase (GCase), and a predisposition to Parkinson’s disease (PD). They demonstrated that increasing GCase activity in human neurons of GD and PD patients can normalize lysosomal function and improve neuronal survival. This research continues at Northwestern University, where Krainc is now Ward Professor and chairman of the department of neurology, and Mazzulli is an assistant professor of neurology.
Compounds that enhance GCase activity represent a potential breakthrough opportunity to develop an entirely new class of agents to treat GD and PD. The risk of developing PD in the broader population has also been firmly linked to GCase mutations. This illustrates how rare diseases can be used as model systems for developing therapeutics for common neurodegenerative disorders.
“This inventive approach to treating Parkinson’s disease was really facilitated by the success of current treatment strategies, such as enzyme replacement therapy, that have enabled the Gaucher disease patient population to live longer,” said Been. “However, an unfortunate consequence of this longevity is the finding that Gaucher patients experience a 20-fold increased risk of developing Parkinson’s disease. At LTI, our lead development program is a completely novel approach to treating Parkinson’s disease by developing small molecules that can cross the blood-brain barrier to increase the activity of the GCase enzyme in the lysosome and potentially reduce Parkinson’s disease risk.”
“This seed funding will be instrumental in enabling LTI to progress its GCase program through the lead-generation process and in setting us up for preclinical development,” said Carpenter, who chairs LTI’s board of directors. “I look forward to working with Kees and his team to refine LTI’s GCase modulator platform over the coming months, with an eye toward advancing breakthrough therapeutics for Parkinson’s disease.”
“We are pleased to support LTI through this seed round of funding,” said Bruce Booth, Ph.D., partner at Atlas Venture. “Atlas believes that LTI’s GCase modulator platform holds great promise as a foundation for discovering therapeutics for neurodegenerative diseases like Parkinson’s, through exploring the implications of known relationships between lysosomal storage and neurodegenerative disorders.”
“Our long-term mission is to develop targeted therapies for children and adults with neurodegenerative disorders that will be based on in-depth, mechanistic understanding of these disorders,” added Krainc.
With this financing, new board members will include Booth; Clay Thorp, general partner at Hatteras Venture Partners; Steve Hall, Ph.D., venture partner at Lilly Ventures; Bernard Davitian, vice president and managing director of Sanofi-Genzyme BioVentures; and Carole Nuechterlein, head of Roche Venture Fund. CEO Been also serves as a member of the board of directors. Nessan Bermingham, Ph.D., venture partner at Atlas Venture, and Reza Halse, Ph.D., partner at Partners Innovation Fund, have been appointed as board observers.
About the Implications of Lysosomal Storage Disorders on Neurodegenerative Diseases
Lysosomal storage disorders (LSDs) are a group of approximately 60 known genetically-inherited diseases characterized by a deficiency of various vital enzymes. All LSDs consist of neurological components, but Gaucher disease (GD) is the most common LSD, occurring when the gene that encodes the lysosomal enzyme glucocerebrosidase (GCase) is mutated and unable to effectively break down its substrate, glucosylceramide. This results in a build-up of lipids in patients’ cells, causing serious health issues.
Recent genetic research suggests that GCase mutations may also cause a predisposition to Parkinson’s disease (PD). The manifestation of the neurotoxic aggregation of the protein alpha-synuclein, also known as Lewy bodies, is the hallmark symptom of PD. Lysosomal Therapeutics Inc.’s (LTI) initial research shows that restoring lysosomal function in human neurons of GD and PD patients may normalize the otherwise-elevated levels of alpha-synuclein. In addition to its work with GD and PD, LTI is investigating other lysosomal enzyme deficiencies and their respective genetic links to common neurodegenerative diseases.
About Lysosomal Therapeutics Inc.
Lysosomal Therapeutics Inc. (LTI) is dedicated to innovative small-molecule research and development in the field of neurodegeneration, yielding new treatment options for patients with severe neurological diseases. Our strategy leverages the clinically-validated link between lysosome-based genetic disorders and neurodegenerative diseases to establish a unique and effective molecular platform for novel drug discovery. LTI’s lead program targets Gaucher-related neurodegeneration, Parkinson’s disease and other synucleinopathies. www.lysosomaltx.com
Company Contact Media Contact
Kees Been Kari Watson
CEO MacDougall Biomedical Communications
(617) 913-0166 (781) 235-3060
Kees.Been@LysosomalTx.com KWatson@MacBioCom.com
# # #
Upper Extremity Muscle Strength Preserved Over 48 Weeks in HIBM Patients on 6 Grams
NOVATO, Calif., April 30, 2014 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (Nasdaq:RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced the presentation of detailed results from a 48-week Phase 2 clinical study of sialic acid extended-release (SA-ER, UX001) tablets in 47 patients with hereditary inclusion body myopathy (HIBM; also known by its new name as GNE myopathy), a rare, progressive muscle-wasting disease. SA-ER is designed to replace the deficient sialic acid substrate in patients with HIBM.
Topline results were previously reported in December 2013, and the following abstract was presented at the Emerging Sciences session of the 66th American Academy of Neurology (AAN) Annual Meeting in Philadelphia.
Z. Argov, Y. Caraco, H. Lau, A. Pestronk, P. Shieh, A. Skrinar, J. Mayhew, J. Martinisi, E. Kakkis
Oral sialic acid extended release (SA-ER) stabilizes upper extremity muscle strength in human GNE myopathy: A Phase 2 study
Patients in the study were initially randomized to receive placebo, 3 grams, or 6 grams of SA-ER per day. After 24 weeks, placebo patients crossed over to either 3 grams or 6 grams total daily dose, on a blinded basis, for an additional 24 weeks. The final analysis compared change at week 48 from baseline for the combined groups at 6 grams versus 3 grams of SA-ER. Assessments included pharmacokinetics, composites of upper extremity and lower extremity muscle strength as measured by dynamometry, other clinical endpoints, patient reported outcomes, and safety.
At 24 weeks, assessments of upper extremity composite of muscle strength showed a statistically significant difference in the 6 gram group compared to placebo (+2.33 kg; 5.5% relative difference from baseline; p=0.040). At 48 weeks, a statistically significant difference between the combined 6 gram group and the combined 3 gram group was observed (+3.44 kg; 8.5% relative difference from baseline; p=0.0033). Patients with less advanced disease (able to walk more than 200 meters at baseline), a predefined subset that comprised approximately 70% of total enrollment, showed a more pronounced difference (+4.69 kg; 9.7% relative difference from baseline; p=0.00055).
"We are encouraged that the preservation of upper extremity muscle strength on 6 grams was sustained compared to a decline on placebo or 3 grams," said Emil D. Kakkis, M.D., Ph.D., Chief Executive Officer of Ultragenyx. "We plan to discuss the Phase 2 and higher dose results with the regulatory authorities to establish the next steps for the SA-ER program."
The lower extremity composite showed a similar pattern of response but did not show a statistically significant difference between the dose groups. None of the groups showed a significant decline during the treatment period. Clinical endpoints related to walking, including the six-minute walk test (6MWT), did not reveal significant differences; there was no significant increase or decline.
The GNE Myopathy Functional Activity Scale (GNEM-FAS), a novel specific patient-reported outcome measure developed to assess the clinical meaningfulness of changes in function, did not show differences at 24 weeks but at 48 weeks showed a positive trend in total (p=0.086), mobility (p=0.087), and upper extremity scores (p=0.096) in the combined 6 gram versus the combined 3 gram groups. An alternative post-hoc statistical analysis using a statistical model incorporating all of the repeated data recorded over time did show statistical significance in these GNEM-FAS outcomes.
SA-ER appeared to be well tolerated with no serious adverse events observed to date in either dose group, and no dosedependent treatment-emergent adverse events were identified. Most adverse events were mild to moderate and most commonly gastrointestinal.
The company continues to treat patients in an extension study evaluating an increased 12 gram daily dosage of sialic acid based on the dose dependence observed at weeks 24 and 48 of the Phase 2 study. Data from the increased dose is expected in late 2014.
About Hereditary Inclusion Body Myopathy
Hereditary inclusion body myopathy (HIBM) is also known as GNE myopathy. HIBM is a rare, severe, progressive, genetic neuromuscular disease caused by a defect in the biosynthetic pathway for sialic acid, with onset in the late teens or twenties. The body's failure to produce enough sialic acid causes muscles to slowly waste away and can lead to very severe disability, with patients typically becoming wheelchair bound and losing most major muscle function within ten to 20 years from onset. There are approximately 1,200 to 2,000 HIBM patients in the developed world, and there is currently no approved therapy.
About Ultragenyx
Ultragenyx is a development-stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with an initial focus on serious, debilitating genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
Forward-Looking Statements
Except for the historical information contained herein, the matters set forth in this press release, including statements regarding timing of release of additional data, parameters of the extension study, Ultragenyx's plans, expectations, goals, objectives, milestones, clinical studies, and product development are forward-looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance, or achievements to differ significantly from those expressed or implied by the forward-looking statements. Such risks and uncertainties include, among others, the uncertainties inherent in the clinical drug development process, including the regulatory approval process, the timing of our regulatory filings, and other matters that could affect the availability or commercial potential of our drug candidate. Ultragenyx undertakes no obligation to update or revise any forward-looking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 24, 2014, and its future periodic reports to be filed with the Securities and Exchange Commission.
CONTACT:
Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
Phase 1b PROMPT Study in Hematologic Malignancies Expected to Begin in Mid-2014
SAN DIEGO, April 23, 2014 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE), a biopharmaceutical company engaged in the discovery and development of adult stem cell modulators to treat orphan diseases, announced today that the U.S. Food and Drug Administration (FDA) has cleared its Investigational New Drug Application (IND) amendment to evaluate PROHEMA® in pediatric patients undergoing hematopoietic stem cell (HSC) transplantation for the treatment of hematologic malignancies. The FDA's clearance of the IND amendment allows the Company to expand its clinical investigation of PROHEMA®, which to date has only been administered to adults, in pediatric patients as young as one year of age. The Company plans to initiate enrollment of the "PROMPT" (PROHEMA® for the treatment of hematologic Malignancies in PediaTric patients undergoing single umbilical cord blood transplantation) study, which is designed to enroll up to 18 patients, between the ages of 1 and 18, at three leading U.S. pediatric transplant centers in mid-2014.
"Each year, over 3,500 children in the U.S. are diagnosed with leukemia, many of whom may ultimately require HSC transplantation," commented Pratik Multani, M.D., M.S., Chief Medical Officer of Fate Therapeutics. "Building upon our encouraging clinical experience with PROHEMA® in adult patients, we look forward to expanding our clinical program to pediatric patients with life-threatening forms of leukemia and other hematologic malignancies. Our goal is to deliver a pharmacologically-optimized HSC therapeutic that can support rapid and durable hematologic and immunologic reconstitution, and enable the curative potential of HSC transplantation in patients across a wide range of ages and a broad spectrum of lifethreatening malignant and rare genetic disorders."
PROHEMA® (16, 16-dimethyl prostaglandin E2, or dmPGE2, modulated cord blood), which is currently being evaluated in a Phase 2 clinical trial in adults, is the Company's lead pharmacologically-modulated HSC therapeutic. In 2010, the FDA granted PROHEMA® orphan designation for the enhancement of stem cell engraftment in patients undergoing allogeneic HSC transplantation. The PROMPT study is an open-label Phase 1b clinical trial of PROHEMA® in pediatric patients undergoing single umbilical cord blood transplantation for the treatment of various hematologic malignancies, such as acute lymphoblastic leukemia (ALL) and acute myeloid leukemia (AML),following myeloablative conditioning. Consistent with the recently initiated Phase 2 clinical trial in adults, the manufacture of PROHEMA® in the PROMPT study will utilize the Company's nutrient-rich media formulation, which has been shown in in vivo preclinical studies to improve HSC viability as well as HSC engraftment by more than two-fold as compared to a standard cell processing media. The primary endpoint of the PROMPT study is safety as assessed by neutrophil engraftment. The study will also evaluate various parameters of efficacy including additional measures of neutrophil engraftment, platelet engraftment, rates of graft failure, acute graft versus host disease and serious infection, and disease-free and overall survival.
About PROHEMA®
PROHEMA® is a pharmacologically-modulated, cord blood-derived hematopoietic stem cell (HSC) therapeutic. PROHEMA® is produced through a proprietary, two-hour, ex vivo cell modulation process that results in rapid activation of key biological pathways involved in homing, proliferation and survival of HSCs. In preclinical testing, PROHEMA® has demonstrated the potential to accelerate engraftment and to drive durable hematopoietic reconstitution, without the need for multi-week expansion protocols. In an initial Phase 1b clinical trial in adult patients with hematologic malignancies undergoing double umbilical cord blood transplant (dUCBT), the median time to neutrophil recovery ( > 500 cells/μL) with PROHEMA® was 17.5 days, which compares favorably to historical norms for patients undergoing dUCBT. In that trial, 100-day survival with PROHEMA® was 100%, and PROHEMA® provided the dominant source of hematopoiesis in 10 of 12 evaluable subjects, suggesting that treatment with PROHEMA® may accelerate engraftment and drive durable and preferential hematologic reconstitution.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the discovery and development of pharmacologic modulators of adult stem cells to treat orphan diseases. The Company utilizes established pharmacologic modalities, including small molecules and therapeutic proteins, and well-characterized biological mechanisms to enhance the therapeutic potential of adult stem cells. The Company has built two adult stem cell modulation platforms: a hematopoietic stem cell (HSC) modulation platform, which seeks to optimize the therapeutic potential of HSCs for treating patients with hematologic malignancies and rare genetic disorders, and a muscle satellite stem cell modulation platform, which seeks to activate the regenerative capacity of muscle for treating patients with degenerative muscle disorders. The Company is presently advancing its lead product candidate, PROHEMA ®, a pharmacologically-modulated HSC therapeutic, in Phase 2 clinical development for hematologic malignancies. Fate Therapeutics is also advancing its proprietary Wnt7a protein analogs in preclinical development for the treatment of muscular dystrophies. Fate Therapeutics is headquartered in San Diego, CA. For more information, please visit www.fatetherapeutics.com.
Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the therapeutic potential of PROHEMA®, and our clinical development plans for PROHEMA®, including the timing of, and our ability to conduct, the PROMPT study. These and any other forward-looking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, the risks that the results of PROHEMA® observed in prior preclinical and clinical development may not be replicated in our PROMPT clinical trial, or other current or subsequent clinical trials, of PROHEMA®, and that PROHEMA® may not produce the therapeutic benefits suggested by the results observed in our prior clinical development, or may cause other unanticipated adverse effects, in current or subsequent clinical trials, the risk of cessation or delay of any ongoing or planned preclinical or clinical development activities for a variety of reasons, including additional information that may be requested or additional obligations that may be imposed by the FDA as a condition to our continuation of clinical trials with PROHEMA®, any difficulties or delays in patient enrollment in the PROMPT study, or any adverse events or other negative results that may be observed in the PROMPT study. For a discussion of other risks and uncertainties, and other important factors, any of which could cause our actual results to differ from those contained in the forward-looking statements, see the risks and uncertainties detailed in the company's periodic filings with the Securities and Exchange Commission, including but not limited to the company's Form 10-K for the fourth quarter and year ended December 31, 2013, and from time to time the company's other investor communications. Fate Therapeutics is providing the information in this press release as of this date and does not undertake any obligation to update any forward-looking statements contained in this press release as a result of new information, future events or otherwise.
CONTACT:
Paul Cox, Stern Investor Relations, Inc.
212.362.1200, paul@sternir.com
SAN DIEGO, April 10, 2014 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE), a biopharmaceutical company engaged in the discovery and development of adult stem cell modulators to treat orphan diseases, announced today that the U.S. Patent and Trademark Office (PTO) has issued Patent No. 8,691,573 entitled "Stem Cell Cultures." The newly issued patent claims a class of small molecule inhibitors of Rho-associated kinase (ROCK) that are crucial to the therapeutic application of human induced pluripotent stem cells (hiPSCs). Modulators belonging to the patented class have been shown to be necessary for the high-throughput derivation of transgene-free hiPSCs, and for the maintenance, survival and genomic stability of hiPSCs in culture. Fate Therapeutics holds an exclusive license from The Scripps Research Institute (TSRI) to the patent in all commercial fields.
"We believe the class of ROCK inhibitors covered by these issued claims is a key component to enable the clinical manufacture of hiPSCs for therapeutic applications," said Scott Wolchko, Chief Operating and Financial Officer of Fate Therapeutics. "These newly issued claims complement the hiPSC-related cell compositions previously patented under our Whitehead Institute for Biomedical Research intellectual property portfolio, which we believe are foundational to hiPSC generation. Our growing patent estate covering small molecule-enhanced reprogramming and pluripotency maintenance and expansion is a reflection of the breadth of our proprietary platform for developing iPSC-based regenerative therapeutics."
Thiazovivin, one specific ROCK inhibitor covered within the patented class, was first discovered by Sheng Ding, Ph.D., a scientific founder of Fate Therapeutics. Under a research collaboration between TSRI and Fate Therapeutics, Dr. Ding and his team first demonstrated that Thiazovivin, in combination with other small molecules, increases the reprogramming efficiency of human fibroblasts into hiPSCs by 200-fold as compared to non-chemically enhanced methods of hiPSC generation (Lin, T., et al, Nature Methods 6, 805 - 808 (2009)). Additional work by both Dr. Ding and Fate Therapeutics further differentiated the ability of Thiazovivin and its related class of molecules, as compared to other ROCK inhibitors, to significantly enhance hiPSC survival and homogeneity. Thiazovivin, as well as compositions and cell culture media comprising Thiazovivin, are patented under U.S. Patent No. 8,044,201 entitled "Stem Cell Cultures," to which Fate Therapeutics holds an exclusive license from TSRI in all commercial fields.
Last month, Fate scientists published an article in the journal Stem Cell Reports describing its proprietary stem cell modulation platform for developing hiPSC-based regenerative therapeutics. The Company's therapeutic platform consists of stage-specific cell culture systems containing combinations of small molecule modulators including ROCK inhibitors, and enables the rapid, parallel derivation of hiPSC clones and their subsequent expansion and survival as transgene-free, single cells in culture. Fate Therapeutics is currently researching therapeutic applications of hiPSC-derived myogenic progenitor cells and hematopoietic cells.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the discovery and development of pharmacologic modulators of adult stem cells to treat orphan diseases. The Company utilizes established pharmacologic modalities, including small molecules and therapeutic proteins, and well-characterized biological mechanisms to enhance the therapeutic potential of adult stem cells. The Company has built two adult stem cell modulation platforms: a hematopoietic stem cell (HSC) modulation platform, which seeks to optimize the therapeutic potential of HSCs for treating patients with hematologic malignancies and rare genetic disorders, and a muscle satellite stem cell modulation platform, which seeks to activate the regenerative capacity of muscle for treating patients with degenerative muscle disorders. The Company is presently advancing its lead product candidate, PROHEMA ®, a pharmacologically-modulated HSC therapeutic, in Phase 2 clinical development for hematologic malignancies. Fate Therapeutics is also advancing its proprietary Wnt7a protein analogs in preclinical development for the treatment of muscular dystrophies. Fate Therapeutics is headquartered in San Diego, CA. For more information, please visit www.fatetherapeutics.com.
Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the therapeutic potential of our programs for the modulation of adult stem cells to treat orphan diseases, as well as the therapeutic potential of our hiPSC platform technology. These and any other forward-looking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, the risks that our hiPSC platform may not enable us to develop hiPSC-derived myogenic progenitor cells or hematopoietic cells suitable for therapeutic applications. For a discussion of other risks and uncertainties, and other important factors, any of which could cause our actual results to differ from those contained in the forward-looking statements, see the risks and uncertainties detailed in the company's periodic filings with the Securities and Exchange Commission, including but not limited to the company's Form 10-K for the fourth quarter and year ended December 31, 2013, and from time to time the company's other investor communications. Fate Therapeutics is providing the information in this press release as of this date and does not undertake any obligation to update any forward-looking statements contained in this press release as a result of new information, future events or otherwise.
CONTACT:
Paul Cox, Stern Investor Relations, Inc.
212.362.1200, paul@sternir.com
Data Presented at American College of Medical Genetics and Genomics (ACMG) Annual Clinical Genetics Meeting
NOVATO, Calif., March 27, 2014 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (Nasdaq:RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced the presentation of preliminary data from the Phase 1/2 study of recombinant human beta-glucuronidase (rhGUS, UX003), an investigational therapy for the treatment of mucopolysaccharidosis 7 (MPS 7, Sly syndrome).
The following abstract is being presented at the ACMG Annual Clinical Genetics Meeting in Nashville, Tennessee.
S. Jones, C. Breen, A. Skrinar, W. Sly, E. Kakkis.
Phase 1/2 Open-Label Study to Assess Safety, Efficacy, and Dose of Recombinant Human
Beta-Glucuronidase (rhGUS) in Subjects with Mucopolysaccharidosis Type VII.
The Phase 1/2 open-label clinical study is assessing the safety, efficacy, and dose of rhGUS administered every other week via intravenous infusion in a 12-week primary analysis phase, which will be followed by dose-exploration and long-term extension.
Preliminary results from three patients who have been administered 2 mg/kg of rhGUS every other week for two, six, and 12 weeks show evidence of clearance of lysosomal storage as indicated by the decline in urinary glycosaminoglycan (GAG) excretion beginning at two weeks of treatment of approximately 30-50%.
At the 12 week assessment of the first patient, absolute liver size was reduced by approximately 11%. This represents a 46% decrease in the excess liver size above normal for age and gender. The remaining patients have not yet reached the 12 week time point for liver size assessment.
No serious adverse events were observed during up to 12 weeks of treatment, and no infusion-associated reactions were observed after a total of 13 infusions to date in these three subjects. The study will continue and additional 12-week interim data are expected in 2014. If the efficacy and safety results for all treated subjects are supportive and the dose confirmed, the company intends to initiate a pivotal Phase 3 study enrolling at least 12 patients in 2014.
About MPS 7
Mucopolysaccharidosis type 7 (MPS 7, Sly syndrome), originally described in 1973 by William Sly, M.D., is a rare genetic, metabolic disorder and is one of 11 different MPS disorders. MPS 7 is caused by the deficiency of beta-glucuronidase, an enzyme required for the breakdown of the glycosaminoglycans (GAGs) dermatan sulfate and heparan sulfate. These complex GAG carbohydrates are a critical component of many tissues. The inability to properly break down GAGs leads to a progressive accumulation in many tissues and results in a multi-system disease.
While its clinical manifestations are similar to MPS 1 and MPS 2, MPS 7 is one of the rarest among the MPS disorders. MPS 7 has a wide spectrum of clinical manifestations and can present as early as at birth. There are no approved therapies for MPS 7 today. The use of enzyme replacement therapy as a potential treatment is based on 20 years of research work in murine models of the disease. Enzyme replacement as a strategy is well established in the MPS field as there are currently four approved enzyme replacement therapies for other MPS disorders: MPS 1 (Aldurazyme®, laronidase), MPS 2 (Elaprase®, idursulfase), MPS 4A (Vimizim™, elosulfase alfa), and MPS 6 (Naglazym®e, galsulfase).
Ultragenyx initiated a Phase 1/2 study in the UK to evaluate the safety, tolerability, efficacy, and dose of intravenous administration of rhGUS in December 2013.
About Ultragenyx
Ultragenyx is a development-stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with an initial focus on serious, debilitating metabolic genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
Forward-Looking Statements
Except for the historical information contained herein, the matters set forth in this press release, including statements regarding Ultragenyx's plans, expectations, goals, objectives, milestones, clinical studies, and product development are forward-looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance, or achievements to differ significantly from those expressed or implied by the forward-looking statements. Such risks and uncertainties include, among others, the uncertainties inherent in the clinical drug development process, including the regulatory approval process, the timing of our regulatory filings, and other matters that could affect the availability or commercial potential of our drug candidate. Ultragenyx undertakes no obligation to update or revise any forward looking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's Annual Report on Form 10-K filed with the Securities and Exchange Commission on March 24, 2014, and its future periodic reports to be filed with the Securities and Exchange Commission.
CONTACT:
Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
NOVATO, CA – March 24, 2014 – Ultragenyx Pharmaceutical Inc. (NASDAQ: RARE), a biopharmaceutical company focused on the development of novel products for rare and ultrarare diseases, today reported its financial results and business highlights for the fourth quarter and year ended December 31, 2013.
“Ultragenyx completed a landmark year in 2013, during which we achieved multiple milestones in our vision to build a next generation rare disease company,” said Emil D. Kakkis, Ph.D., M.D., Chief Executive Officer and President of Ultragenyx. “We have expanded our pipeline to five clinical programs in Phase 1/2 or Phase 2 studies, and with the completion of our initial public offering in early 2014, we believe we are well‐positioned to advance multiple programs in parallel to address the significant unmet medical needs of these patients.”
Fourth Quarter and Full Year 2013 Financial Results
For the fourth quarter of 2013, Ultragenyx reported a net loss attributable to common stockholders of $18.7 million, or $4.98 per share, basic and diluted, compared with a net loss attributable to common stockholders for the fourth quarter of 2012 of $6.8 million, or $2.83 per share, basic and diluted.
For the year ended December 31, 2013, Ultragenyx reported a net loss attributable to common stockholders of $50.3 million, or $14.87 per share, basic and diluted, compared with a net loss attributable to common stockholders for the year ended December 31, 2012 of $19.6 million, or $14.20 per share, basic and diluted.
Total operating expenses for 2013 were $32.3 million compared with $16.0 million for 2012. Total operating expenses for the fourth quarter of 2013 were $9.5 million compared with $4.7 million for the same period in 2012. The increase in total operating expenses is due to the addition of triheptanoin and KRN23 to Ultragenyx’s clinical development pipeline, advancements in the development of sialic acid extended release (SA‐ER) and recombinant human beta‐glucuronidase (rhGUS), and increased headcount to support the company’s growth.
Cash, cash equivalents, and short‐term investments were $53.4 million as of December 31, 2013. Based on current operating levels, the company expects that existing cash, cash equivalents and short‐term investments, including approximately $121.7 million in net proceeds received from the initial public offering in February 2014, will be sufficient to fund operations into 2016.
Recent Highlights
KRN23 anti‐FGF23 Monoclonal Antibody in X‐linked Hypophosphatemia (XLH)
- Results from the Phase 1 single dose study in 38 adult XLH patients were presented at the American Society for Bone and Mineral Research Annual Meeting in October 2013 and published in the Journal of Clinical Investigation in February 20141. The data demonstrated that KRN23 was well tolerated and increased serum phosphate and vitamin D levels compared to placebo at higher doses.
rhGUS in Mucopolysaccharidosis 7 (MPS 7)
- In December 2013, we initiated a Phase 1/2 study to evaluate the safety, tolerability, efficacy, and dose of rhGUS in up to five MPS 7 patients between five and 30 years of age.
- In February 2014, results from the treatment of a single patient under an emergency investigational new drug (eIND) application were presented at the Lysosomal Disease Network’s 10th Annual World Symposium. The preliminary data showed a reduction in lysosomal storage based on reduced excretion of urinary glycosaminoglycans and a reduction in the size of the enlarged liver and spleen. The patient showed an improvement of pulmonary function and no infusion‐associated reactions during the first 14 weeks of treatment.
Triheptanoin in Long‐Chain Fatty Acid Oxidation Disorders (LC‐FAOD)
- In February 2014, we initiated a Phase 2 study in approximately 30 severely affected LCFAOD patients. A principal goal of the study is to determine the appropriate clinical endpoints and patient population for testing in potential later‐stage pivotal studies.
- Multiple investigator‐sponsored and compassionate use trials testing triheptanoin in LCFAOD patients are also ongoing.
Triheptanoin in Glucose Transporter Type‐1 Deficiency Syndrome (Glut1 DS)
- In March 2014, we initiated a randomized, double‐blind, placebo‐controlled, parallel‐group Phase 2 study in up to 50 Glut1 DS patients between three and 17 years of age inclusive who are currently not fully compliant with ketogenic diet and continue to have seizures. The primary efficacy objective is the reduction in frequency of seizures compared to placebo.
- Multiple investigator‐sponsored and compassionate use trials in Glut1 DS as well as in other indications are also ongoing.
SA‐ER in Hereditary Inclusion Body Myopathy (HIBM)
- In December 2013, we released top‐line results after 48 weeks of treatment from the Phase 2 randomized study of SA‐ER in 47 HIBM patients. The data showed that a modest increase in upper extremity muscle strength composite at the higher dose compared to a decline in the lower dose group was statistically significant. A positive trend was seen in a patientreported outcome of functional activity. The results were consistent with the 24‐week analysis. SA‐ER appeared to be well tolerated with no serious adverse events observed to date in either dose group.
- We continue to treat patients in an extension study evaluating an increased daily dosage of sialic acid based on the dose dependence observed at weeks 24 and 48 of the Phase 2 study.
Corporate Highlights
- In February 2014, we closed our initial public offering of 6,624,423 shares of common stock at an initial public offering price of $21.00 per share, which includes the exercise in full by the underwriters of their option to purchase up to an additional 864,054 additional shares of common stock. Net proceeds from the offering were approximately $121.7 million, after deducting underwriting discounts and commissions, estimated offering expenses, and a dividend paid to preferred stockholders.
- In January 2014, concurrent with the pricing of our initial public offering, we appointed Clay B. Siegall, Ph.D. and Matthew K. Fust to our board of directors. Dr. Siegall is President, Chief Executive Officer, and Chairman of the Board of Seattle Genetics, Inc. Mr. Fust is former Executive Vice President and Chief Financial Officer of Onyx Pharmaceuticals, Inc.
Anticipated 2014 Program Milestones
KRN23 in XLH
- Release data from the Phase 1/2 adult repeat‐dose studies completed by our collaborative partner Kyowa Hakko Kirin Co., Ltd. (KHK).
- Initiate a Phase 2 study in pediatric XLH patients following discussions with multiple regulatory agencies on our pediatric study design.
rhGUS in MPS 7
- Release early urinary glycosaminoglycans data on the first three MPS 7 patients dosed with rhGUS in our Phase 1/2 study in March 2014 at the Annual Clinical Genetics Meeting (ACMG).
- Release interim 12‐week data from our Phase 1/2 study of rhGUS in patients with MPS 7 in the second half of 2014.
- Initiate a pivotal Phase 3 study of at least 12 patients in the second half of 2014 if the 12‐ week Phase 1/2 data are positive.
Triheptanoin in LC‐FAOD
- Continue to enroll and treat patients in the ongoing Phase 2 study of triheptanoin in approximately 30 severely affected LC‐FAOD patients. We expect that data from this study should be available in 2015.
Triheptanoin in Glut1 DS
- Continue to enroll and treat patients in the ongoing Phase 2 study of triheptanoin in up to 50 Glut1 DS patients. We expect to release data from this trial in 2015.
SA‐ER in HIBM
- Present detailed data from the 48‐week randomized, controlled Phase 2 study of SA‐ER in 47 HIBM patients at the American Academy of Neurology (AAN) Annual Meeting in April. The abstract was one of ten chosen for presentation in a late‐breaking news session called the Emerging Sciences session. A brief topline presentation of the results will take place on April 30, 2014 at approximately 6:30pm ET and will be followed by a poster session.
- Release data from the ongoing extension study of SA‐ER at a higher daily dosage in late 2014.
About Ultragenyx
Ultragenyx is a development‐stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra‐rare diseases, with an initial focus on serious, debilitating metabolic genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx’s strategy is predicated upon time and cost‐efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company’s website at www.ultragenyx.com.
1Carpenter TO, et al. Randomized trial of the anti‐FGF23 antibody KRN23 in X‐linked hypophosphatemia. J Clin Invest. 2014. doi:10.1172/JCI72829.
Forward‐Looking Statements
Except for the historical information contained herein, the matters set forth in this press release, including statements regarding Ultragenyx's plans, potential opportunities, expectations, projections, goals, objectives, milestones, strategies, product pipeline, clinical studies, product development, release of data and the potential benefits of its products under development are forward‐looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward‐looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance or achievements to differ significantly from those expressed or implied by the forward‐looking statements. Such risks and uncertainties include, among others, the uncertainties inherent in the clinical drug development process, including the regulatory approval process, the timing of our regulatory filings and other matters that could affect the availability or commercial potential of our drug candidates. Ultragenyx undertakes no obligation to update or revise any forward‐looking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward‐looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's prospectus filed with the Securities and Exchange Commission on January 31, 2014, and its future periodic reports to be filed with the Securities and Exchange Commission.
Ultragenyx Pharmaceutical Inc.
March 18, 2014, Seattle, WA and South San Francisco, CA – Immune Design, a clinical-stage immunotherapy company focused on the development of novel immune-based therapies for cancer and other chronic conditions, today announced the appointment of Franklin M. Berger, CFA to the company’s board of directors.
Mr. Berger is a major player in the biotechnology industry with more than 25 years of experience in capital markets and financial analysis. He has served as a consultant in the biotechnology industry since 2003, advising major biopharmaceutical firms, mid-capitalization biotechnology companies, specialist asset managers and venture capital companies in a range of areas including business development, strategic advisory/financings, partnering and royalty acquisition. Prior to that, Mr. Berger spent 12 years in sell-side equity research, most recently as Managing Director, U.S. Equity Research and Senior Biotechnology Analyst at J. P. Morgan Securities, Inc.
“Franklin brings a wealth of experience to Immune Design,” said Carlos Paya, M.D., Ph.D., President and Chief Executive Officer at Immune Design. “He is a great addition to our experienced board and will provide vital strategic financial insights and advice to help guide the continued growth of our exciting company.”
During his five years at J. P. Morgan Securities, Inc., Mr. Berger was involved with the issuance of over $12 billion in biotechnology company equity or equity-linked securities. He was associated with several notable financings in the biotechnology sector, including the Genentech IPO and the initial large Celgene Corporation financings. Mr. Berger began his career as a sell-side analyst at Josephthal & Co. in 1991, subsequently moving to Salomon Smith Barney in 1997 serving as Director, Equity Research and Senior Biotechnology Analyst. Institutional Investor Magazine ranked him on J. P. Morgan’s All-Star Research Team. The Wall Street Journal (WSJ) selected Mr. Berger as the No. 1 ranked biotechnology analyst in its All-Star Analyst Survey in 1997 and No. 2 ranked in the WSJ’s 2000 Survey. In 2000, he became a Founding Fellow of the Biotechnology Study Center at New York University School of Medicine. Mr. Berger currently serves on the board of four public biotechnology companies: Seattle Genetics, Inc., Five Prime Therapeutics, Inc., Bellus Health, Inc. and BioTime, Inc. He also serves or has served on private biotech company boards including Caprion Proteomics (sold in July 2012), Inc. and ViroChem Pharma, Inc., which was purchased by Vertex Pharmaceuticals, Inc. for $400 million in 2009.
Mr. Berger holds an M.B.A. from Harvard Business School and an M.A. in International Economics and a B.A. in International Relations from Johns Hopkins University.
“The field of cancer immunotherapy is attracting notable interest and investment, and Immune Design is very well-positioned to take a leadership role in this evolving landscape through application of its novel discovery platforms that have generated multiple product candidates now in clinical trials,” said Mr. Berger. “I look forward to working with the Immune Design team.”
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing leading-edge technologies that target dendritic cells in vivo to generate and expand strong cytotoxic T cells (CTLs) for the treatment of cancer and other chronic diseases. The company has a multi-faceted set of clinical programs on going that are the product of its two synergistic discovery platforms: DCVexTM, a novel lentiviral vector platform engineered to deliver antigen-encoding nucleic acids directly to dendritic cells in vivo; and GLAASTM, a TLR4-agonist platform that activates dendritic cells by up-regulating key molecules for efficient antigen presentation and produces Th1 cytokines to enhance the immune response. Immune Design has offices in Seattle, Washington and South San Francisco, California. For more information, visit www.immunedesign.com.
# # #
Contact:
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
First Patient Enrolled in PUMA Study
Approved for Conduct at Ten Leading U.S. Centers
Interim Data Expected in 2H14
SAN DIEGO, March 12, 2014 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE), a biopharmaceutical company engaged in the discovery and development of adult stem cell modulators to treat orphan diseases, announced today the enrollment of the first patient in its "PUMA" (PROHEMA® in UMbilical cord blood transplant in Adults) study, a Phase 2 clinical trial of PROHEMA® (16, 16-dimethyl prostaglandin E2, or dmPGE2, modulated cord blood) using the Company's nutrient-rich media formulation. The PUMA study is designed to assess the efficacy and safety of PROHEMA® in a randomized, controlled setting in patients undergoing hematopoietic stem cell (HSC) transplantation for the treatment of hematologic malignancies. The trial has been approved for conduct at ten major HSC transplant centers in the United States. Safety reviews are planned after six and 12 subjects, respectively, have been treated with PROHEMA® in the PUMA study, and the Company intends to provide a clinical update following the completion of these reviews. Full data on the primary efficacy endpoint are expected in mid-2015.
"Despite important advancements in the pharmacotherapy of blood cancers, many patients with leukemia or lymphoma advance to a stage where hematopoietic stem cell transplantation represents the only curative therapeutic option," commented Christian Weyer, M.D., M.A.S., President and Chief Executive Officer of Fate Therapeutics. "PROHEMA® is representative of Fate's commitment to developing stem cell therapeutics to address significant unmet medical needs and offer life-saving therapeutic solutions to patients with hematologic malignancies and rare genetic disorders. We have worked diligently to optimize the clinical potency and efficacy profile of PROHEMA®, and we are excited to pursue clinical development of PROHEMA® in both malignant and non-malignant disorders."
During 2013, scientists at Fate Therapeutics demonstrated that the therapeutic profile of PROHEMA® may be further enhanced, as compared to its previous clinical use, by incorporating the Company's nutrient-rich media (NRM) formulation into the manufacture of PROHEMA®. In in vivo preclinical studies, PROHEMA® manufactured using its NRM formulation exhibited improved HSC viability and a more than two-fold improvement in HSC engraftment as compared to a standard cell processing media used previously in the clinical development of PROHEMA®. The Company has worked closely with the FDA over the past nine months to enable the clinical manufacture of PROHEMA® using its NRM formulation for the PUMA study.
"While allogeneic HSC transplantation is a proven therapeutic intervention with curative potential for patients with hematologic malignancies and over 50 rare genetic disorders, it is associated with significant treatment-related limitations and 100-day mortality rates between 20% to 30%," said Corey Cutler M.D., M.P.H., F.R.C.P.C., Associate Professor of Medicine at the Dana- Farber Cancer Institute and Harvard Medical School and principal investigator in the PUMA trial and the Company's prior clinical trials of PROHEMA®. "Engraftment, particularly as measured by time to the engraftment of neutrophils, is correlated with key clinical outcomes of HSC transplantation including the length of hospital stays, rates of serious infections and overall treatment-related morbidity and mortality. I applaud Fate Therapeutics for its dedication and effort in bringing the most optimal product candidate to patients."
The PUMA study is an open-label, randomized, controlled, multi-center Phase 2 clinical trial designed to enroll 60 subjects, age 15 to 65 years, and will use the Company's NRM formulation in the manufacture of PROHEMA®. Patients will be randomized at a ratio of 2:1, with approximately 40 patients receiving PROHEMA® plus an unmanipulated cord blood unit and approximately 20 patients receiving two unmanipulated cord blood units. At the discretion of the treating physician, patients will be conditioned using an intensive myeloablative or a reduced intensity regimen. The primary endpoint of the PUMA study is the cumulative incidence of time to neutrophil engraftment as compared to a pre-specified control median, which will be adjusted based upon the median time to neutrophil engraftment calculated for the patients enrolled in the control arm. Secondary endpoints include additional measures of neutrophil and platelet engraftment, rates of graft failure, acute graft versus host disease and serious infection, and disease-free and overall survival.
About PROHEMA®
PROHEMA® is a pharmacologically-modulated, cord blood-derived hematopoietic stem cell (HSC) therapeutic. PROHEMA® is produced through a proprietary, two-hour, ex vivo cell modulation process that results in rapid activation of key biological pathways involved in homing, proliferation and survival of HSCs. In preclinical testing, PROHEMA® has demonstrated the potential to accelerate engraftment and to drive durable hematopoietic reconstitution, without the need for multi-week expansion protocols. In an initial Phase 1b clinical trial in adult patients with hematologic malignancies undergoing double umbilical cord blood transplant (dUCBT), the median time to neutrophil recovery ( > 500 cells/μL) with PROHEMA® was 17.5 days, which compares favorably to historical norms for patients undergoing dUCBT. In that trial, 100-day survival with PROHEMA® was 100%, and PROHEMA® provided the dominant source of hematopoiesis in 10 of 12 evaluable subjects, suggesting that treatment with PROHEMA® may accelerate engraftment and drive durable and preferential hematologic reconstitution.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the discovery and development of pharmacologic modulators of adult stem cells to treat orphan diseases, including certain hematologic malignancies, lysosomal storage disorders and muscular dystrophies. The Company utilizes established pharmacologic modalities, including small molecules and therapeutic proteins, and well-characterized biological mechanisms to enhance the therapeutic potential of adult stem cells. The Company has built two adult stem cell modulation platforms: a hematopoietic stem cell (HSC) modulation platform, which seeks to optimize the therapeutic potential of HSCs for treating patients with hematologic malignancies and rare genetic disorders that are undergoing hematopoietic stem cell transplantation, and a muscle satellite stem cell modulation platform, which seeks to activate the regenerative capacity of muscle for treating patients with degenerative muscle disorders. The Company is presently advancing its lead product candidate, PROHEMA®, a pharmacologically-modulated HSC therapeutic derived from umbilical cord blood, in Phase 2 clinical development for hematologic malignancies. Fate Therapeutics is also advancing its proprietary Wnt7a protein analogs in preclinical development for the treatment of muscular dystrophies. Fate Therapeutics is headquartered in San Diego, CA. For more information, please visit www.fatetherapeutics.com.
Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the therapeutic potential of PROHEMA®, and our clinical development plans for PROHEMA®, including the timing of, and our ability to conduct, safety reviews of subjects in the PUMA study, and the timing and availability of both interim and full data in the trial. These and any other forward-looking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, the risk of cessation or delay of any ongoing or planned preclinical or clinical development activities for a variety of reasons, including additional information that may be requested or additional obligations that may be imposed by the FDA as a condition to our continuation of clinical trials with PROHEMA®, any difficulties or delays in patient enrollment in the PUMA study, and any adverse events or other negative results that may be observed in the PUMA study. For a discussion of other risks and uncertainties, and other important factors, any of which could cause our actual results to differ from those contained in the forward-looking statements, see the risks and uncertainties detailed in the company's periodic filings with the Securities and Exchange Commission, including but not limited to the company's Form 10-Q for the quarter ended September 30, 2013, and from time to time the company's other investor communications. Fate Therapeutics is providing the information in this press release as of this date and does not undertake any obligation to update any forward-looking statements contained in this press release as a result of new information, future events or otherwise.
CONTACT:
Paul Cox, Stern Investor Relations, Inc.
212.362.1200, paul@sternir.com
Novato, CA, March 11, 2014 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (NASDAQ: RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, announced the first patient enrolled in the Phase 2 study of triheptanoin (UX007) for the treatment of glucose transporter type-1 deficiency syndrome (Glut1 DS), at Columbia University. Glut1 DS, also known as De Vivo disease, is a rare, severely debilitating disease characterized by seizures, developmental delay, and movement disorder.
"Glut1 deficiency was first described by us at the Columbia University Medical Center in 1991, so we are pleased to have the opportunity to enroll the first patient in the triheptanoin clinical trial sponsored by Ultragenyx. It is clear that more effective symptomatic and disease-modifying treatments are needed for this rare disease, and we hope this trial is the next step towards that ultimate goal," said Dr. Darryl De Vivo, the Sidney Carter Professor of Neurology and Professor of Pediatrics at Columbia University Medical Center in New York City.
The global, randomized, double-blind, placebo-controlled, parallel-group Phase 2 clinical trial will evaluate safety and efficacy in up to 50 Glut1 DS patients between 3 and 17 years of age inclusive, who are currently not on or not fully compliant with ketogenic diet and continue to have seizures. The primary efficacy objective is the reduction in frequency of seizures compared to placebo following a 6-week baseline period and subsequent 8-week placebo-controlled treatment period. The blinded treatment period will be followed by an open-label extension period in which all patients will be treated with triheptanoin through week 52. Targeted enrollment may be modified based on an interim analysis. We anticipate that data from this trial will be available in 2015.
"We are advancing triheptanoin for Glut1 DS based on its anticipated ability to provide an alternative source of energy to the brain that could result in seizure control and improvement in other aspects of the disease," commented Emil D. Kakkis, M.D., Ph.D., Ultragenyx's Chief Executive Officer. "The placebo-controlled Phase 2 study is a significant step in the evaluation of triheptanoin for this rare and devastating disease."
About Glut1 DS and Triheptanoin
Glut1 DS is a severely debilitating disease characterized by seizures, developmental delay, and movement disorder. Glut1 DS is caused by a mutation in the gene encoding the Glut1 protein, which is responsible for transporting glucose across the bloodbrain barrier. Because glucose is the primary source of energy for the brain, this disorder results in a chronic state of energy deficiency in the brain. Glut1 DS is a rare disease. Studies suggest a range of 3,000 to 7,000 Glut1 DS patients in the United States. There are currently no approved treatments specific to Glut1 DS.
Triheptanoin is a specially designed synthetic triglyceride compound intended to provide patients with the medium-length, oddchain fatty acid, heptanoate. Heptanoate can circulate and also be further metabolized to four- and five-carbon ketone bodies in the liver. All of these metabolites are able to cross the blood-brain barrier without using the deficient Glut1 transporter, and can provide an alternative energy source for the brain when glucose is limited. Heptanoate also crosses the blood-brain barrier and can be converted to glucose.
About Ultragenyx
Ultragenyx is a development-stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with an initial focus on serious, debilitating metabolic genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
Forward-Looking Statements
Except for the historical information contained herein, the matters set forth in this press release, including statements regarding Ultragenyx's plans, potential opportunities, expectations, projections, goals, objectives, milestones, strategies, product pipeline, clinical studies, product development and the potential benefits of its products under development are forward-looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance or achievements to differ significantly from those expressed or implied by the forward-looking statements. Such risks and uncertainties include, among others, the uncertainties inherent in the clinical drug development process, including the regulatory approval process, the timing of our regulatory filings and other matters that could affect the availability or commercial potential of our drug candidate. Ultragenyx undertakes no obligation to update or revise any forwardlooking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's prospectus filed with the Securities and Exchange Commission on January 31, 2014, and its future periodic reports to be filed with the Securities and Exchange Commission.
CONTACT:
Contact Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
March 6, 2014, Seattle, WA and South San Francisco, CA – Immune Design, a clinical-stage immunotherapy company focused on the development of novel immune-based therapies for cancer and other chronic conditions, today announced the appointment of biotechnology and pharmaceutical industry veteran William (“Bill”) R. Ringo to the company’s board of directors.
Mr. Ringo brings more than four decades of biopharmaceutical experience in a variety of leadership and advisory roles. He currently serves as a senior advisor with Barclays Capital, the global investment banking division of Barclays Bank PLC, and as a strategic advisor with California-based Sofinnova Ventures, whose investment focus includes life sciences startup companies in the United States and Europe. Prior to this, Mr. Ringo served as Senior Vice President of Strategy and Business Development for Pfizer, where he led the company’s overall strategic planning and business development efforts until his retirement in 2010.
“Bill’s breadth and depth of experience in the biopharmaceutical industry will be invaluable to Immune Design,” said Carlos Paya, M.D., Ph.D., President and Chief Executive Officer at Immune Design. “We are pleased to have him join our distinguished board of directors and know he will be a helpful advisor as we continue to mature our immunotherapy product pipeline.”
Mr. Ringo’s experience includes nearly 30 years with Eli Lilly and Company, where he served in multiple executive roles. Mr. Ringo also has served in numerous leadership roles within the biotechnology industry. From 2004 to 2006, he served as President and Chief Executive Officer of Abgenix, which focused on developing human antibodies as agents to treat cancer and other diseases. Amgen purchased the company in 2006 for $2.2 billion. From 2001 to 2007, he served on various boards of directors, including Encysive Pharmaceuticals, Inc., Inspire Pharmaceuticals, Inc. and InterMune, Inc. where he was the non-executive chairman of the Board of Directors after serving as interim Chief Executive Officer from June to September 2003. Mr. Ringo also served on the Onyx Pharmaceuticals board of directors from 2010 until 2013. Mr. Ringo currently serves as chairman of Sangamo BioSciences and a board member for Ascendis Medical Technology and BioCrossroads. Mr. Ringo earned a B.S. in Business Administration and an M.B.A. from the University of Dayton.
“Immune Design is developing an impressive pipeline of in vivo cancer immunotherapeutics through the application of its innovative discovery platforms,” said Mr. Ringo. “The company has distinct clinical approaches that may result in multiple meaningful therapeutics. I’m pleased to be joining the board of directors and look forward to assisting the team in advancing this next-generation strategy.”
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing leading-edge technologies that target dendritic cells in vivo to generate and expand strong cytotoxic T cells (CTLs) for the treatment of cancer and other chronic diseases. The company has a multi- faceted set of clinical programs on going that are the product of its two synergistic discovery platforms: DCVexTM, a novel lentiviral vector platform engineered to deliver antigen-encoding nucleic acids directly to dendritic cells in vivo; and GLAASTM, a TLR4-agonist platform that activates dendritic cells by up-regulating key molecules for efficient antigen presentation and produces Th1 cytokines to enhance the immune response. Immune Design has offices in Seattle, Washington and South San Francisco, California. For more information, visit www.immunedesign.com.
# # #
Contact:
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206-769-9219
SAN DIEGO, March 6, 2014 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE), a biopharmaceutical company engaged in the discovery and development of adult stem cell modulators to treat orphan diseases, announced today the publication of an article in the journal Stem Cell Reports by Fate scientists demonstrating high-throughput derivation of human induced pluripotent stem cells (hiPSCs) that exhibit characteristics necessary for therapeutic application. The publication describes the use of the Company's hiPSC platform, consisting of stage-specific cell culture systems, to enable rapid, parallel derivation of hiPSC clones and their subsequent expansion as transgene-free, single cells in culture. The Company's proprietary combinations of small molecule modulators, which include ROCK, GSK3 and MEK pathway inhibitors, used in the culture systems were found to be critical in promoting characteristics of the ground state of pluripotency including pluripotent culture stability, homogeneity and survival.
"This publication further highlights the scientific and technological progress we have made in defining the most stable and efficient platform for hiPSC-based cell therapy applications," commented Peter Flynn Ph.D., Senior Vice President, Early Program Development at Fate Therapeutics. "While there are multiple approaches to cellular reprogramming, we are dedicated to deriving hiPSCs with pluripotent qualities necessary for robust, reproducible hiPSC generation and expansion. We believe that the use of our proprietary small molecule inhibitors is critical for enabling the development of potentially transformative, hiPSC-based regenerative therapeutics."
In January of this year, Fate scientists published a separate article in Stem Cells Translation Medicine reporting the efficient differentiation of hiPSCs to a homogeneous population of skeletal muscle cells using the Company's proprietary hiPSC platform. In support of the Company's muscle regeneration therapeutic program, Fate scientists sourced fibroblasts from healthy volunteers as well as from patients with Duchenne and Becker muscular dystrophies, reprogrammed the fibroblasts to hiPSCs, and then differentiated the hiPSCs to muscle myoblasts with extremely high efficiency. The hiPSC-derived skeletal muscle cells closely resembled primary muscle cells, and were functionally responsive to treatment with the hypertrophic proteins insulin-like growth factor 1 (IGF-1) and Wnt7a. Fate Therapeutics is currently developing proprietary Wnt7a protein analogs for the treatment of muscle-related diseases and disorders. In addition, the Company is currently researching therapeutic applications of hiPSC-derived myogenic progenitor cells and hematopoietic cells.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the discovery and development of pharmacologic modulators of adult stem cells to treat orphan diseases, including certain hematologic malignancies, lysosomal storage disorders and muscular dystrophies. The Company utilizes established pharmacologic modalities, including small molecules and therapeutic proteins, and well-characterized biological mechanisms to enhance the therapeutic potential of adult stem cells. The Company has built two adult stem cell modulation platforms: a hematopoietic stem cell (HSC) modulation platform, which seeks to optimize the therapeutic potential of HSCs for treating patients with hematologic malignancies and rare genetic disorders that are undergoing hematopoietic stem cell transplantation, and a muscle satellite stem cell modulation platform, which seeks to activate the regenerative capacity of muscle for treating patients with degenerative muscle disorders. The Company is presently advancing its lead product candidate, PROHEMA®, a pharmacologically-modulated HSC therapeutic derived from umbilical cord blood, in Phase 2 clinical development for hematologic malignancies. Fate Therapeutics is also advancing its proprietary Wnt7a protein analogs in preclinical development for the treatment of muscular dystrophies. Fate Therapeutics is headquartered in San Diego, CA. For more information, please visit www.fatetherapeutics.com.
Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the therapeutic potential of our programs for the modulation of adult stem cells to treat orphan diseases, including PROHEMA® and our Wnt7a protein analogs, our preclinical and clinical development plans, including our ability to resume enrollment of the PROHEMA-03 trial and our ability to initiate IND-enabling activities for our Wnt7a-analog program, and the impact of our hiPSC platform technology. These and any other forward-looking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, the risk of cessation or delay of any ongoing or planned preclinical or clinical development activities for a variety of reasons, including additional information that may be requested or additional obligations that may be imposed by the FDA as a condition to our resumption or continuation of the PROHEMA-03 trial, any inability to complete the cell-line development, in vivo studies, and pharmacokinetic and toxicology assessments necessary to support further IND-enabling activities and advance our Wnt7a analog program into clinical development, and any inability to develop hiPSCs, and hiPSC-derived cells, suitable for cell-therapy based applications. For a discussion of other risks and uncertainties, and other important factors, any of which could cause our actual results to differ from those contained in the forward-looking statements, see the risks and uncertainties detailed in the company's periodic filings with the Securities and Exchange Commission, including but not limited to the company's Form 10-Q for the quarter ended September 30, 2013, and from time to time the company's other investor communications. Fate Therapeutics is providing the information in this press
release as of this date and does not undertake any obligation to update any forward-looking statements contained in this press release as a result of new information, future events or otherwise.
CONTACT:
Paul Cox, Stern Investor Relations, Inc.
212.362.1200, paul@sternir.com
Trial to evaluate antigen-specific, active immunotherapy approach
February 27, 2014, Seattle, WA and San Francisco, CA – Immune Design, a clinical-stage immunotherapy company focused on the development of novel immune-based therapies for cancer and other chronic conditions, today announced treatment of the first patient in a Phase 1 clinical trial of ID-G305, a cancer immunotherapy investigational agent from the company’s GLAASTM platform.
The Phase 1 open label, dose-escalation trial is designed to evaluate the safety, tolerability and immunogenicity of ID-G305 in patients with unresectable, relapsed, or metastatic cancer expressing the NY-ESO-1 antigen, including melanoma, sarcoma, lung, ovarian or breast cancer. The trial is being conducted in up to 18 patients at several clinical centers in the United States.
“The development of agents that harness the patient’s cellular immune system to kill tumor cells is a promising approach for the treatment of human malignancies,” said Amit Mahipal, M.D., M.P.H., Medical Director, Clinical Research Unit, Moffitt Cancer Center, and a Principal Investigator in the study. “We look forward to evaluating this novel therapeutic candidate, ID-G305, and its ability to generate immune responses in cancer patients.”
“We are pleased to launch this first clinical trial of ID-G305, which has been designed to stimulate multiple arms of the immune system,” said Richard Kenney, M.D., Chief Medical Officer of Immune Design. “The adjuvant component, GLA-SE, is a unique, potent, clinical-stage toll-like receptor-4 (TLR4) agonist that has demonstrated favorable clinical safety in multiple clinical trials to date. Combining this with the targeted NY-ESO-1 protein offers the potential to generate effective immune activity in cancer patients.”
About ID-G305
ID-G305 triggers a potent and specific cellular and humoral anti-tumor response, as well as stimulating the innate immune response. This product candidate was generated from Immune Design’s GLAASTM discovery platform, which leverages a proprietary synthetic small molecule adjuvant glucopyranosyl lipid A (GLA), a novel TLR4 agonist. ID-G305 is part of Immune Design’s “Specific Antigen” approach, which drives the in vivo generation of a strong, antigen-specific CTL response against selected antigens present in the tumor. ID-G305 consists of recombinant NY-ESO-1 protein, which is expressed by a variety of cancers, formulated with GLA-SE. The NY-ESO-1 protein is provided by the Ludwig Cancer Research and Cancer Research Institute with which the company has a collaboration to advance cancer immunotherapy research.
GLA has been evaluated in multiple clinical studies involving nearly 1,000 patients to date, where it has been shown to be well tolerated.
About Immune Design
Immune Design is a clinical-stage immunotherapy company employing leading-edge technologies that target in vivo dendritic cells to generate and expand strong cytotoxic T cells (CTLs) for the treatment of cancer and other chronic diseases. The company’s clinical programs are the product of its two synergistic discovery platforms: DCVexTM, a novel lentiviral vector platform engineered to deliver antigen-encoding nucleic acids directly to dendritic cells in vivo, and GLAASTM, a TLR4-agonist platform that activates dendritic cells by up-regulating key molecules for efficient antigen presentation and produces Th1 cytokines to enhance the immune response. Immune Design has offices in Seattle, Washington and South San Francisco, California. For more information, visit www.immunedesign.com.
# # #
Contact:
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206.769.9219
SAN DIEGO, Feb. 26, 2014 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE), a biopharmaceutical company engaged in the discovery and development of adult stem cell modulators to treat orphan diseases, announced today the release of new data on the observed effects of ex vivo pharmacologic modulation on CD8+ T cells and immune reconstitution from its previously-completed Phase 1b clinical trial of PROHEMA® (16, 16-dimethyl prostaglandin E2, or dmPGE2, modulated cord blood) in adult patients undergoing hematopoietic stem cell (HSC) transplantation for hematologic malignancy (the ProHema-01 trial). Researchers affiliated with Harvard Medical School performed an analysis of the CD8+ T cell compartment using patient samples from the ProHema-01 trial. At Day 100, subjects who received PROHEMA® and an unmanipulated cord blood unit (n=12) showed a two-fold increase in the percentage of naïve and early memory T cell fraction within the CD8+ T cell compartment as compared to subjects who received two unmanipulated cord blood units (n=9). These naïve and early memory
CD8+ T cell populations are believed to play a key role in promoting immune reconstitution and viral immunity following cord blood transplantation. These findings were recently published in Blood Cancer Journal (Li, L. et. al. Blood Cancer Journal 2014).
"In addition to the well-established effect of dmPGE2 on enhancing the engraftment properties of hematopoietic stem cells, our molecular characterization and immunological assessments provide new evidence that dmPGE2 can also significantly improve the functional properties of T cells contained within human umbilical cord blood," stated Dr. Vicki Boussiotis, M.D., Ph.D., Professor, Department of Medicine, Harvard Medical School, and senior author on the Blood Cancer Journal publication. "Enhancing the survival and immunological properties of T cells, and in particular those of naïve CD8+ T cells, may have significant therapeutic implications for harnessing immune memory in the context of both pathogen- and tumor-specific immunity."
Consistent with these reported immunomodulatory effects, low rates of viral reactivation were observed in the ProHema-01 trial. Specifically, cytomegalovirus (CMV) reactivation occurred in only two of 12 PROHEMA® subjects (17%), with no cases of CMV disease. In addition, no cases of Epstein-Barr virus (EBV)-associated post-transplant lymphoproliferative disorders (PTLD) were observed. These observations compare favorably to rates of CMV reactivation and EBV-associated PTLD reported in the literature of 36-56% and up to 16%, respectively.
"The functional properties of both hematopoietic stem cells and T cells are critical determinants of patient outcomes following allogeneic umbilical cord blood transplantation," commented Pratik Multani, M.D., M.S., Chief Medical Officer of Fate Therapeutics. "These novel scientific and clinical observations raise the possibility that T cell-related mechanisms may also be enhanced using ex vivo pharmacologic modulation strategies. We look forward to evaluating patient outcomes related to dmPGE2-mediated immunomodulatory effects in our planned Phase 2 clinical trial of PROHEMA®."
PROHEMA® is currently in clinical development for use in adult patients with hematologic malignancies undergoing double umbilical cord blood transplantation.Following an optimization of the PROHEMA® manufacturing process, the Company expects to resume enrollment of its Phase 2 clinical trial (the ProHema-03 Trial) in the first half of 2014, with the goal of generating full data on the primary and major secondary endpoints related to engraftment in mid-2015.
About PROHEMA®
PROHEMA® is a pharmacologically-modulated, cord blood-derived hematopoietic stem cell (HSC) therapeutic. PROHEMA® is produced through a proprietary, two-hour, ex vivo cell modulation process that results in rapid activation of key biological pathways involved in homing, proliferation and survival of HSCs. In preclinical testing, PROHEMA® has demonstrated the potential to accelerate engraftment and to drive durable hematopoietic reconstitution, without the need for multi-week expansion protocols. In an initial Phase 1b study in adult patients with hematologic malignancies undergoing double umbilical cord blood transplant (dUCBT), the median time to neutrophil recovery ( > 500 cells/μL) with PROHEMA® was 17.5 days, which compares favorably to historical norms for patients undergoing dUCBT. In that study, 100-day survival with PROHEMA® was 100%, and PROHEMA® provided the dominant source of hematopoiesis in 10 of 12 evaluable subjects, suggesting that treatment with PROHEMA® may accelerate engraftment and drive durable and preferential reconstitution.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the discovery and development of pharmacologic modulators of adult stem cells to treat orphan diseases, including certain hematologic malignancies, lysosomal storage disorders and muscular dystrophies. The Company utilizes established pharmacologic modalities, including small molecules and therapeutic proteins, and well-characterized biological mechanisms to enhance the therapeutic potential of adult stem cells. The Company has built two adult stem cell modulation platforms: a hematopoietic stem cell (HSC) modulation platform, which seeks to optimize the therapeutic potential of HSCs for treating patients with hematologic malignancies and rare genetic disorders that are undergoing hematopoietic stem cell transplantation, and a muscle satellite stem cell modulation platform, which seeks to activate the regenerative capacity of muscle for treating patients with degenerative muscle disorders. The Company is presently advancing its lead product candidate, PROHEMA®, a pharmacologically-modulated HSC therapeutic derived from umbilical cord blood, which is in Phase 2 clinical development for hematologic malignancies. Fate Therapeutics is also advancing its proprietary Wnt7a protein analogs in preclinical development for the treatment of muscular dystrophies. Fate Therapeutics is headquartered in San Diego, CA. For more information, please visit www.fatetherapeutics.com.
Forward-Looking Statements
This release contains "forward-looking statements" within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the therapeutic potential of our programs for the modulation of adult stem cells to treat orphan diseases, the ability of dmPGE2 to improve the functional properties of T cells within human umbilical cord blood, the ability of ex vivo pharmacologic modulation of adult stem cells to enhance T cell-related mechanisms, the potential therapeutic benefits of enhancing the survival and immunological properties of T cells, our ability to resume enrollment of the ProHema-03 trial in the first half of 2014 following an optimization of the PROHEMA® manufacturing process, and our ability to generate full data on the primary and major secondary endpoints related to engraftment in mid-2015. These and any other forward-looking statements in this release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by
such forward-looking statements. These risks and uncertainties include, but are not limited to, the risks that the results observed in our ProHema-01 trial may not be replicated in our ProHema-03 trial or other subsequent clinical trials of PROHEMA®, PROHEMA® may not produce the therapeutic benefits suggested by the results observed in our ProHema-01 trial or may cause other unanticipated adverse effects in subsequent clinical trials, or we may cease or delay any of our ongoing or planned preclinical or clinical development activities for a variety of reasons, including additional information that may be requested or additional obligations that may be imposed by the FDA as a condition to our resumption or continuation of the ProHema-03 trial, any inability to obtain an adequate clinical supply of PROHEMA®, any delays in enrollment of our ProHema- 03 trial, and any negative results following resumption of the ProHema-03 trial. For a discussion of other risks and uncertainties, and other important factors, any of which could cause our actual results to differ from those contained in the forward-looking statements, see the risks and uncertainties detailed in the company's periodic filings with the Securities and Exchange Commission, including but not limited to the company's Form 10-Q for the quarter ended September 30, 2013, and from time to time the company's other investor communications. Fate Therapeutics is providing the information in this press release as of this date and does not undertake any obligation to update any forward-looking statements contained in this press release as a result of new information, future events or otherwise.
CONTACT:
Paul Cox, Stern Investor Relations, Inc.
212.362.1200, paul@sternir.com
Open-Label Study to Assess Safety and Clinical Effects of Triheptanoin
NOVATO, Calif., Feb. 11, 2014 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (Nasdaq:RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced that the first patient has been enrolled in an open-label Phase 2 study to assess safety and clinical effects of triheptanoin, also known as UX007, in patients severely affected by long-chain fatty acid oxidation disorders (LC-FAOD). LC-FAOD are a group of autosomal recessive genetic disorders characterized by metabolic deficiencies in which the body is unable to break down and convert long chain fatty acids into energy.
"We look forward to studying triheptanoin in this prospective clinical study and taking another step towards determining the potential role for triheptanoin in helping patients with LC-FAOD to better manage their debilitating disease," said Emil D. Kakkis, M.D., Ph.D., Chief Executive Officer and President of Ultragenyx. "The goal for this study is to determine the optimal patient population and endpoints for evaluation in a potential future Phase 3 clinical trial."
The prospective, interventional, open-label Phase 2 study will evaluate triheptanoin treatment in approximately 30 severely affected LC-FAOD patients, ages 6 months to 35 years, exhibiting significant clinical manifestations of LC-FAOD despite current therapy. Prior to initiating treatment with triheptanoin, subjects will continue their current therapy for four weeks to establish their baseline condition. Triheptanoin will then be titrated to an expected target dose of 25-35% of total daily caloric intake via oral administration, while ensuring tolerability. The patients will be followed to evaluate the effects of triheptanoin treatment over 24 weeks, then may continue treatment for an additional 54 weeks. The study will assess the impact of triheptanoin on several endpoints, including cycle ergometer performance, 12-minute walk test, muscle strength, creatine kinase levels, hypoglycemia, liver size, and cardiac disease. The study will be conducted at approximately eight clinical sites in the United States and Europe.
During the treatment period, the primary objective of the study is to evaluate the impact of triheptanoin on acute clinical pathophysiology associated with LC-FAOD, while the secondary objectives of the study are to evaluate the safety of triheptanoin treatment and its effects on energy metabolism in LC-FAOD patients. The objective of the extension period of the study is to evaluate the impact of triheptanoin on major clinical events, including hospitalizations, emergency room visits, and emergency interventions associated with LC-FAOD.
About LC-FAOD and Triheptanoin
LC-FAOD are a group of autosomal recessive genetic disorders characterized by metabolic deficiencies in which the body is unable to convert long-chain fatty acids into energy. It is estimated that 2,000 to 3,500 patients are afflicted with LC-FAOD in the US, where fatty acid oxidation disorders are now detected by newborn screening. There are six main genetic diseases that cause LC-FAOD, and the main clinical program is focused on the most common four forms: carnitine palmitoyltransferase II (CPT-II) deficiency, long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency, trifunctional protein deficiency (TFP), and very long-chain acyl-CoA dehydrogenase (VLCAD) deficiency.
LC-FAOD patients are currently treated by the avoidance of fasting, low-fat/high carbohydrate diets, carnitine supplementation and medium chain triglyceride (MCT) oil. Despite current therapy, many patients still have significant metabolic events including hospitalizations and significantly increased mortality due to LC-FAOD; a mortality rate of more than 50% has been observed in spite of treatment with current therapy.
Triheptanoin, also known as UX007, is a purified form of a specially designed synthetic triglyceride compound. Triheptanoin is intended to provide patients with medium-length, odd-chain fatty acids that are metabolized to replace intermediate substrates in fatty acid oxidation downstream of their genetic block in fatty acid metabolism. Triheptanoin is also metabolized to a substrate that replaces deficient intermediates in the tricarboxylic acid (TCA cycle), a key energy-generating process. Triheptanoin can also support production of glucose and glycogen (gluconeogenesis). Together, the substrates produced by triheptanoin during metabolism are intended to improve energy production in FAOD patients.
About Ultragenyx
Ultragenyx is a development-stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with an initial focus on serious, debilitating metabolic genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
Forward-Looking Statements
Except for the historical information contained herein, the matters set forth in this press release, including statements regarding Ultragenyx's plans, potential opportunities, expectations, projections, goals, objectives, milestones, strategies, product pipeline, clinical studies, product development and the potential benefits of its products under development are forward-looking statements within the meaning of the "safe harbor" provisions of the Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve substantial risks and uncertainties that could cause our clinical development programs, future results, performance or achievements to differ significantly from those expressed or implied by the forward-looking statements. Such risks and uncertainties include, among others, the uncertainties inherent in the clinical drug development process, including the regulatory approval process, the timing of our regulatory filings and other matters that could affect the availability or commercial potential of our drug candidate. Ultragenyx undertakes no obligation to update or revise any forwardlooking statements. For a further description of the risks and uncertainties that could cause actual results to differ from those expressed in these forward-looking statements, as well as risks relating to the business of the Company in general, see Ultragenyx's prospectus filed with the Securities and Exchange Commission on January 31, 2014, and its future periodic reports to be filed with the Securities and Exchange Commission.
CONTACT:
Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
NOVATO, Calif., Feb. 6, 2014 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc. (Nasdaq:RARE), a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced the appointment of Clay B. Siegall, Ph.D. and Matthew Fust to the company's Board of Directors effective January 30, 2014. Dr. Siegall, currently President, Chief Executive Officer and Chairman of the Board of Seattle Genetics, Inc., and Mr. Fust, formerly Executive Vice President and Chief Financial Officer of Onyx Pharmaceuticals, Inc. will serve as independent directors for Ultragenyx. With these additions, Ultragenyx extends gratitude and appreciation for the counsel provided by Mr. Ben Auspitz, who has resigned from his position on the Board effective January 30, 2014.
"On behalf of the Board of Directors and Executive Team at Ultragenyx, we enthusiastically welcome Clay and Matt to the Board. Both Clay and Matt are proven leaders in the biopharmaceutical industry. Their broad expertise and diverse experience will be of enormous value to Ultragenyx as it enters the next phase of its development," said Eran Nadav, Ph.D., Managing Director at TPG Biotech and Chairman of the Board of Ultragenyx. "We would also like to acknowledge Ben Auspitz for his invaluable contributions to Ultragenyx over the past several years."
"Ultragenyx has made substantial progress with its clinical programs over the past several years," Dr. Siegall commented. "I look forward to sharing my experience towards that progress to help achieve success for Ultragenyx and especially for patients affected by rare and ultra-rare diseases."
"It is exciting to be part of a company that is not only patient-focused but leveraging an innovative approach to addressing severe and potentially life-threatening diseases," said Mr. Fust. "I look forward to working closely with the Board and the Ultragenyx team to deliver on this endeavor."
Dr. Siegall currently serves as President, Chief Executive Officer and Chairman of the Board of Seattle Genetics, Inc., a biotechnology company focused on the development and commercialization of innovative antibody-based therapies for the treatment of cancer. Dr. Siegall cofounded Seattle Genetics in 1998. Prior to Seattle Genetics, Dr. Siegall worked for the Bristol-Myers Squibb Pharmaceutical Research Institute from 1991 to 1997 and the National Cancer Institute, National Institutes of Health from 1988 to 1991. In addition to Seattle Genetics, Dr. Siegall serves as a director of Alder BioPharmaceuticals, Inc. and Mirna Therapeutics, Inc., both privately-held biotechnology companies. Dr. Siegall received a B.S. in Zoology from the University of Maryland and a Ph.D. in Genetics from George Washington University.
Mr. Fust was Executive Vice President and Chief Financial Officer of Onyx Pharmaceuticals, Inc., a biopharmaceutical company, from January 2009 until its recent acquisition by Amgen, Inc. From May 2003 to December 2008, Mr. Fust served as Chief Financial Officer at Jazz Pharmaceuticals, Inc., a specialty pharmaceutical company. From 2002 to 2003, Mr. Fust served as Chief Financial Officer at Perlegen Sciences, a biopharmaceutical company. Previously, he was Senior Vice President and Chief Financial Officer at ALZA Corporation, a pharmaceutical company, where he was an executive from 1996 until 2002. From 1991 until 1996, Mr. Fust was a member of the Healthcare Strategy practice at Andersen Consulting. Mr. Fust serves on the Board of Directors of Sunesis Pharmaceuticals, Inc., a biopharmaceutical company. Mr. Fust received a B.A. from the
University of Minnesota and an M.B.A. from the Stanford University Graduate School of Business.
With their addition, Dr. Siegall will become a member of the Compensation Committee, and Mr. Fust will become a member and chair of each of the Audit Committee and the Nominating and Corporate Governance Committee.
About Ultragenyx
Ultragenyx is a development-stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with an initial focus on serious, debilitating metabolic genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
CONTACT:
Ultragenyx Pharmaceutical Inc.
844-758-7273
For Media, Bee Nguyen
For Investors, Robert Anstey
NOVATO, Calif., Jan. 30, 2014 (GLOBE NEWSWIRE) -- Ultragenyx Pharmaceutical Inc., a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced the pricing of its initial public offering of 5,760,369 shares of its common stock at an initial public offering price of $21.00 per share, before underwriting discounts. The shares are expected to begin trading on The NASDAQ Global Select Market on January 31, 2014 under the ticker symbol "RARE." All of the shares of common stock are being offered by Ultragenyx. In addition, Ultragenyx has granted the underwriters a 30-day option to purchase up to an additional 864,054 shares of common stock to cover over-allotments, if any.
Ultragenyx expects to close its initial public offering on February 5, 2014, subject to satisfaction of customary closing conditions. J.P. Morgan Securities LLC and Morgan Stanley & Co. LLC are acting as the joint book-running managers for the offering. Cowen and Company, LLC is acting as lead manager, and Canaccord Genuity Inc. is acting as co-manager. Ropes & Gray LLP served as legal advisor to Ultragenyx, and Latham & Watkins LLP advised the underwriters.
A registration statement relating to these securities was declared effective by the Securities and Exchange Commission on January 30, 2014. Copies of the registration statement, as amended, can be accessed through the SEC's website at www.sec.gov. This offering is being made solely by means of a prospectus, copies of which may be obtained from J.P. Morgan Securities LLC, Attention: Broadridge Financial Solutions, 1155 Long Island Avenue, Edgewood, NY 11717, or from Morgan Stanley & Co. LLC, Attention: Prospectus Department, 180 Varick Street, 2nd Floor, New York, NY 10014.
This press release shall not constitute an offer to sell or the solicitation of an offer to buy, nor shall there be any sale of these securities in any state or jurisdiction in which such offer, solicitation, or sale would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction.
About Ultragenyx
Ultragenyx is a development-stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with an initial focus on serious, debilitating metabolic genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx's strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company's website at www.ultragenyx.com.
CONTACT:
Ultragenyx Pharmaceutical Inc.
415-483-8800
For Media, Bee Nguyen
For Investors, Robert Anstey
Trial to evaluate ID-G100 in patients with Merkel cell carcinoma
January 27, 2014, Seattle, WA and South San Francisco, CA – Immune Design, a clinical-stage biotechnology company focused on the development of novel immune-based therapies for cancer and other chronic conditions, today announced treatment of the first patient in a Phase 1 clinical trial of ID-G100 in patients with Merkel cell carcinoma (MCC).
“This trial will provide insights into the ability of ID-G100 to stimulate an immune response in patients with Merkel cell carcinoma,” said Shailender Bhatia, M.D., medical oncologist at Seattle Cancer Care Alliance, assistant professor, medical oncology division at the University of Washington School of Medicine and principal investigator. “New treatments are greatly needed for this aggressive disease, and we look forward to evaluating this novel immunotherapy approach.”
The Phase 1 open label trial is designed to evaluate the safety, feasibility, clinical efficacy and immunogenicity of ID-G100 in patients with metastatic or logoregional MCC. The trial is being conducted at the University of Washington Medical Center and Seattle Cancer Care Alliance, with support from the Life Sciences Discovery Fund.
“Merkel cell carcinoma is an aggressive cutaneous neuroendocrine carcinoma with few definitive treatment options. We hope this novel therapy will eventually provide these patients with a meaningful new treatment opportunity for this orphan disease,” said Richard Kenney, M.D., Chief Medical Officer of Immune Design. “A core component of ID-G100, GLA, has been evaluated as a molecular vaccine adjuvant in more than 1,000 subjects and has demonstrated the ability to stimulate the innate and adaptive immune system while being well tolerated. The recent discovery of a polyoma virus that is associated with MCC supports the potential for an immunotherapeutic approach.”
About ID-G100
ID-G100 is an investigational agent that includes GLA (Glucopyranosyl Lipid A, a synthetic, Toll-like Receptor-4 agonist) and is a product of the company’s GLAASTM discovery platform. ID-G100 is intended for intra-tumoral injection and is part of Immune Design’s “Endogenous Antigen” approach to treating cancer, which leverages an intratumoral activation of dendritic cells in the context of the tumor’s preexisting broad set of antigens to create a robust local and systemic anti-tumor immune response. Preclinical and clinical data have demonstrated the ability of GLA to significantly activate dendritic cells in animal models and to increase antigen dependent humoral and cellular TH1 immune responses.
Immune Design’s “Specific Antigen” approach, in contrast, delivers specific tumor antigens directly to cancer patients’ dendritic cells using a cutting edge delivery vector specific for a subset of skin dendritic cells. The company is pursuing this approach simultaneously in its ID-G305 and ID-LV305 clinical programs.
For Patients
More information about this study, including additional eligibility criteria and contact information, can be found on clinicaltrials.gov.
About Merkel cell carcinoma
Merkel cell carcinoma is a rare aggressive form of skin cancer that most often occurs on the face, neck and back. The majority of cases are associated with the Merkel cell polyomavirus. Approximately 1,500 new cases of Merkel cell carcinoma are reported in the United States each year and the incidence of the disease has almost tripled in the past two decades. Common treatment options include surgery, chemotherapy and radiation therapy. For more information about Merkel cell carcinoma and available treatment options visit www.merkelcell.org.
About the Life Sciences Discovery Fund
The Life Sciences Discovery Fund (LSDF) was established in 2005 by the Governor and Legislature of Washington to foster growth of the state’s life sciences sector and improve the health and economic wellbeing of its residents. LSDF invests monies from the Master Tobacco Settlement Agreement in research and development across Washington that demonstrate the strongest potential for delivering health and economic returns to the state. To date, LSDF has produced a 7:1 return on Washington’s investment, including over $445 million in additional funding and more than $67 million in health-care cost savings. For more information about LSDF and its grant making, visit: www.lsdfa.org.
About Seattle Cancer Care Alliance
Seattle Cancer Care Alliance is a cancer treatment center that unites doctors from Fred Hutchinson Cancer Research Center, UW Medicine and Seattle Children’s. Our goal, every day, is to turn cancer patients into cancer survivors. Our purpose is to lead the world in the prevention and treatment of cancer. SCCA has five clinical care sites: an outpatient clinic on the Hutchinson Center campus, a pediatric inpatient unit at Seattle Children’s, an adult inpatient unit at UW Medical Center, a medical oncology clinic at EvergreenHealth, and medical and radiation oncology clinics at UW Medicine / Northwest Hospital & Medical Center. Additionally, proton therapy services are provided at SCCA Proton Therapy, A Procure Center. For more information about SCCA, visit www.seattlecca.org.
About Immune Design
Immune Design is a clinical-stage biotechnology company employing leading-edge technologies that target dendritic cells for a more precise immune response for the treatment of cancer and other chronic conditions. The company’s clinical programs are the product of its two synergistic discovery platforms: DCVexTM, a novel lentiviral vector platform engineered to deliver antigen-encoding nucleic acids directly to dendritic cells in vivo, and GLAASTM, a TLR4-agonist platform that activates dendritic cells by up-regulating key molecules for efficient antigen presentation and produces Th1 cytokines to enhance the immune response. Immune Design has offices in
Seattle, Washington and South San Francisco, California. For more information, visit www.immunedesign.com.
# # #
Contact:
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206.769.9219
GAITHERSBURG, Md, January 9, 2014-- GlycoMimetics, Inc. today announced the pricing of its initial public offering of 7,000,000 shares of common stock at a price to the public of $8.00 per share. The shares are scheduled to begin trading on the NASDAQ Global Market under the ticker symbol "GLYC" on January 10, 2014. All of the common stock is being offered by GlycoMimetics. In addition, GlycoMimetics has granted the underwriters a 30-day option to purchase up to 1,050,000 additional shares of common stock from GlycoMimetics. The offering is expected to close on January 15, 2014, subject to customary closing conditions.
Jefferies LLC and Barclays Capital Inc. are acting as joint book-running managers for the offering. Stifel is acting as co-lead manager and Canaccord Genuity Inc. is acting as co-manager.
A registration statement relating to these securities was declared effective by the Securities and Exchange Commission on January 9, 2014.
The offering is being made only by means of a prospectus. A copy of the final prospectus relating to these securities will be filed with the SEC and may be obtained, when available, from Jefferies LLC, Equity Syndicate Prospectus Department, 520 Madison Avenue, 12th Floor, New York, NY 10022, by email at Prospectus_Department@Jefferies.com or by phone at 877- 547-6340 or Barclays Capital Inc., c/o Broadridge Financial Solutions, 1155 Long Island Avenue, Edgewood, NY 11717, by email at Barclaysprospectus@broadridge.com or by phone at 888-603-5847.
This press release shall not constitute an offer to sell or a solicitation of an offer to buy, nor shall there be any sale of these securities in any state or jurisdiction in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction.
About GlycoMimetics, Inc.
GlycoMimetics is a clinical stage biotechnology company focused on the discovery and development of novel glycomimetic drugs to address unmet medical needs resulting from diseases in which carbohydrate biology plays a key role. Glycomimetics are molecules that mimic the structure of carbohydrates involved in important biological processes. Using its expertise in carbohydrate chemistry and knowledge of carbohydrate biology, GlycoMimetics is developing a pipeline of glycomimetic drug candidates that inhibit disease-related functions of carbohydrates, such as the roles they play in inflammation, cancer and infection.
- Investment & Company Information
- Finance
- initial public offering
Contact:
GlycoMimetics, Inc.
Brian Hahn, 240-243-1207
bhahn@glycomimetics.com
Contact Ultragenyx Pharmaceutical Inc.
415-483-8800
For Media, Bee Nguyen
For Investor, Robert Anstey
FOR IMMEDIATE RELEASE:
Ultragenyx Announces Results from Phase 2 Study of Sialic Acid Extended-Release Treatment in Hereditary Inclusion Body Myopathy
Upper extremity muscle strength composite shows statistically significant difference between the 6 gram and 3 gram dose groups
Novato, CA—December 20, 2013—Ultragenyx Pharmaceutical Inc., a biopharmaceutical company focused on the development of novel products for rare and ultra-rare diseases, today announced topline results from a 48-week Phase 2 clinical study of sialic acid extended-release (SA-ER, UX001) tablets in 46 patients with hereditary inclusion body myopathy (HIBM), a progressive muscle-wasting disease. SA-ER is designed to replace the deficient sialic acid substrate in patients with HIBM. Patients were initially randomized to either receive placebo, 3 grams or 6 grams of SA-ER per day. After 24 weeks, placebo patients crossed over to either 3 grams or 6 grams total daily dose, on a blinded basis, for an additional 24 weeks. The final analysis compared change at week 48 from baseline for the combined groups at 6 grams versus 3 grams of SA-ER.
Similar to the results observed at the 24-week interim analysis, at 48 weeks the comparison of the upper extremity composite of muscle strength for the combined group of patients on 6 grams showed a modest increase and a statistically significant difference relative to the decline in strength observed in the combined groups on 3 grams. In the 6-gram cohort treated for 48 weeks, the modest increase in upper extremity strength observed at 24 weeks was sustained relative to a further decline in the comparable 3-gram group. These changes were more pronounced in those patients that have less advanced disease as assessed by a greater walking ability at baseline, a predefined subset. The lower extremity composite did not show a statistically significant difference between the dose groups, but neither group showed a significant decline during the treatment period. The upper extremity and lower extremity composites are predefined endpoints that aggregate change in muscle strength across multiple individually measured muscle groups.
The GNE Myopathy Functional Activity Scale, a patient-reported outcome measure developed to assess the clinical meaningfulness of changes in function, showed a trend toward a positive effect in the combined 6-gram group as compared to the combined 3-gram group. The sit-to-stand test also showed a trend toward a positive effect. Other clinical endpoints related to walking, including the 6 minute walk test did not reveal significant changes in function, upward or downward. SA-ER appeared to be well tolerated with no serious adverse events observed to date in either dose group. Most adverse events were mild to moderate and most commonly gastrointestinal.
“These 48 week data suggest that 6 grams per day of SA-ER is mitigating the normally expected decline in upper extremity muscle strength,” said Emil Kakkis, M.D., Ph.D., Chief Executive Officer of Ultragenyx. “The maintenance of this effect from the 24-week data is encouraging. Given the difference between the dose groups and good safety profile, we plan to test an even higher dose of sialic acid in these patients who have no other approved treatment options.”
The data from the 48-week analysis are expected to be presented in full at a scientific conference in 2014. All 46 patients from the original study have elected to continue into the extension study. The company plans to treat patients with an increased dosage of sialic acid based on the dose-dependence observed at week 48. The first patient has already been treated with a higher dose of sialic acid, and data from the increased dose is expected in late 2014.
About Hereditary Inclusion Body Myopathy
Hereditary inclusion body myopathy (HIBM) is also known as GNE myopathy. HIBM is a severe, progressive, genetic neuromuscular disease caused by a defect in the biosynthetic pathway for sialic acid, with onset in the late teens or twenties. The body’s failure to produce enough sialic acid causes muscles to slowly waste away and can lead to very severe disability, with patients typically becoming wheelchair bound within ten to 20 years from onset. There are approximately 1,200 to 2,000 HIBM patients in the developed world, and there is currently no approved therapy.
About Ultragenyx
Ultragenyx is a privately held, clinical-stage biotechnology company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with an initial focus on serious, debilitating metabolic genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx’s strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company’s website at www.ultragenyx.com.
Contact Ultragenyx Pharmaceutical Inc.
415-483-8800
For Media, Bee Nguyen
For Investors, Robert Anstey
NOVATO, CA – December 4, 2013 – Ultragenyx Pharmaceutical Inc., a biotechnology company, today announced the dosing of the first patient in a Phase 1/2 study of recombinant human beta-glucuronidase (rhGUS, UX003) for MPS 7, in Manchester, UK. MPS 7 is an ultra-rare autosomal recessive lysosomal storage disorder characterized by a deficiency of the enzyme beta-glucuronidase that results in a severe multi-system disease.
William Sly, MD, Chairman Emeritus of the Department of Biochemistry at Saint Louis University commented, “Patients, families, and researchers have been waiting many years for this advancement in the treatment of MPS 7. The initiation of clinical testing of rhGUS is the culmination of decades of work.”
The Phase 1/2 open-label clinical trial will assess the safety and efficacy of rhGUS in a 12-week primary analysis phase, followed by dose-exploration and long-term extension. Five patients between 5 and 30 years of age inclusive will be enrolled and administered rhGUS every other week via intravenous infusion. Interim data from the Phase 1/2 study is expected in 2014.
“MPS 7 is a devastating condition that has been neglected by the drug development community despite 20 years of extensive nonclinical research led by Dr. Sly and his colleagues,” said Emil D. Kakkis, MD, PhD, Chief Executive Officer of Ultragenyx. “The initiation of this Phase 1/2 study is an important milestone for Ultragenyx and for patients with MPS 7.”
About MPS 7
Mucopolysaccharidosis type 7 (MPS 7), originally described in 1973 by William Sly, MD, is a rare genetic, metabolic disorder and is one of 11 different MPS disorders. MPS 7 is caused by the deficiency of beta-glucuronidase, an enzyme required for the breakdown of the glycosaminoglycans (GAGs) dermatan sulfate and heparan sulfate. These complex GAG carbohydrates are a critical component of many tissues. The inability to properly break down GAGs leads to a progressive accumulation in many tissues and results in a multi-system disease.
While its clinical manifestations are similar to MPS 1 and MPS 2, MPS 7 is one of the rarest among the MPS disorders. MPS 7 has a wide spectrum of clinical manifestations and can present as early as at birth. There are no approved therapies for MPS 7 today. The use of enzyme replacement therapy as a potential treatment is based on 20 years of research work in murine models of the disease. Enzyme replacement as a strategy is well established in the MPS field as there are currently three approved enzyme replacement therapies for other MPS disorders: MPS 1 (Aldurazyme®, laronidase), MPS 2 (Elaprase®, idursulfase), and MPS 6 (Naglazyme®, galsulfase).
About Ultragenyx
Ultragenyx is a privately held, development-stage biopharmaceutical company committed to bringing to market novel products for the treatment of rare and ultra-rare diseases, with an initial focus on serious, debilitating metabolic genetic diseases. Founded in 2010, the company has rapidly built a diverse portfolio of product candidates with the potential to address diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no approved therapies.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx’s strategy is predicated upon time and cost-efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company’s website at www.ultragenyx.com.
SOUTH SAN FRANCISCO, Calif., Oct. 30, 2013 /PRNewswire/ -- KaloBios Pharmaceuticals, Inc. (Nasdaq: KBIO) today announced that the U.S. Food & Drug Administration (FDA) has granted the company orphan drug designation for KB001-A, an anti-PcrV monoclonal antibody (mAb) fragment, for the treatment of cystic fibrosis (CF) patients with Pseudomonas aeruginosa (Pa).
(Logo: http://photos.prnewswire.com/prnh/20130225/MM66380LOGO)
KaloBios is currently enrolling patients in a Phase 2 multiple-dose, randomized, double-blind, placebo-controlled clinical trial with KB001-A in CF patients chronically infected with Pa. This 180-patient study is intended to evaluate the efficacy and safety of repeat doses of KB001-A. The primary endpoint is time-to-need for antibiotics. To learn more about this study, including eligibility criteria, please visit http://clinicaltrials.gov/ct2/show/NCT01695343?term=kb001a&rank=1
"The FDA's designation of KB001-A as an orphan drug is another milestone for KaloBios as we continue the clinical
development work required for potential FDA approval," said David W. Pritchard, KaloBios' President and Chief Executive Officer. "We are working hard to complete enrollment of our Phase 2 trial and look forward to evaluating and reporting on the activity of KB001-A in late 2014."
Orphan drug designation is granted by the FDA Office of Orphan Products Development to novel drugs or biologics that treat a rare disease or condition affecting fewer than 200,000 patients in the United States. The designation provides the drug developer with a seven-year period of U.S. marketing exclusivity if the drug is the first of its type approved for the specified indication or if it demonstrates superior safety, efficacy, or a major contribution to patient care versus another drug of its type that was previously granted the designation for the same indication. Orphan designation also provides tax credits for clinical research costs, the ability to apply for annual grant funding, clinical research trial design assistance and waiver of Prescription Drug User Fee Act (PDUFA) filing fees.
About KaloBios
KaloBios Pharmaceuticals, Inc. is developing a portfolio of proprietary, patient-targeted, first-in-class monoclonal antibodies designed to treat severe life-threatening or debilitating diseases for which there is an unmet medical need, with a clinical focus on severe respiratory diseases and cancer.
Currently, KaloBios has three drug development programs:
1. KB003, an anti-GM-CSF mAb with potential to treat inflammatory diseases, being developed for the treatment of severe asthma. Enrollment of 160 patients has been completed in a Phase 2 study in the United States, Europe and Australia.
2. KB001-A, an anti-PcrV mAb fragment that is partnered exclusively with Sanofi Pasteur and is being developed for the prevention and treatment of Pa infections. KaloBios has retained rights for the CF indication and has initiated a 180 patient Phase 2 study in CF subjects with chronic Pa lung infection in the United States. KaloBios has received Orphan Drug designation from both the FDA and the European Commission for KB001-A for the treatment of Pa lung infection in CF. Sanofi is pursuing a ventilator-associated pneumonia prevention indication in the intensive care setting, an indication which has received U.S. FDA Fast Track Designation.
3.KB004, an anti-EphA3 mAb, has potential in treating hematologic malignancies and solid tumors. KaloBios is currently testing this drug in a Phase 1 study in subjects with hematologic malignancies.
All of the company's antibodies were generated using its proprietary Humaneered® technology, a method that converts nonhuman antibodies (typically mouse) into recombinant antibodies that have a high binding affinity to their target and are designed for chronic therapeutic use. The company believes that antibodies produced using its Humaneered® technology offer important clinical and economic advantages over antibodies generated by other methods in terms of high binding affinity, high manufacturing yields, and minimal to no immunogenicity (inappropriate immune response) upon repeat administration in humans.
For more information on KaloBios Pharmaceuticals, please visit our web site at http://www.kalobios.com.
Forward Looking Statements
This release contains forward-looking statements made pursuant to the safe harbor provisions of the Private Securities Litigation Reform Act of 1995, including: the statements under the heading "Anticipated Upcoming Milestones for 2013-2014"; and statements regarding the company's clinical development of KB001-A, KB003 and KB004. Forward-looking statements reflect management's current knowledge, assumptions, judgment and expectations regarding future performance or events. Although management believes that the expectations reflected in such statements are reasonable, they give no assurance that such expectations will prove to be correct and you should be aware that actual results could differ materially from those contained in the forward-looking statements. Forward-looking statements are subject to a number of risks and uncertainties including, but not limited to, the company's limited cash reserves and its ability to obtain additional capital on acceptable terms, or at all, including the additional capital which will be necessary to complete the clinical trials that the company has initiated or plans to initiate; the company's dependence on Sanofi Pasteur for the development and commercialization of KB001-A; the company's ability to successfully complete further development of its programs; the uncertainties inherent in clinical testing, including time to enroll clinical studies; the timing, cost and uncertainty of obtaining regulatory approvals; the company's ability to protect the company's intellectual property; competition; changes in the regulatory landscape or the imposition of regulations that affect the company's products; and other factors listed under "Risk Factors" in the company's Annual Report on Form 10-K filed with the Securities and Exchange Commission on April 1, 2013, the quarterly reports on Form 10-Q filed on May 14, 2013 and August 19, 2013, and the company's other filings with the Securities and Exchange Commission.
All forward-looking statements are expressly qualified in their entirety by this cautionary notice. You are cautioned not to place undue reliance on any forward-looking statements, which speak only as of the date of this release. The company has no obligation, and expressly disclaims any obligation to update, revise or correct any of the forward-looking statements, whether as a result of new information, future events or otherwise.
For more information, visit http://www.kalobios.com.
Contact:
Herb Cross
Chief Financial Officer
KaloBios Pharmaceuticals, Inc.
(650) 243-3114
ir@kalobios.com
Media Contact:
Joan E. Kureczka
Kureczka/Martin Associates
Tel: (415) 821-2413
Mobile: (415) 690-0210
Joan@Kureczka-Martin.com
SOURCE KaloBios Pharmaceuticals, Inc.
News Provided by Acquire Media
October 30, 2013, Seattle, WA and San Francisco, CA – Immune Design, a biotechnology company focused on the development of novel immune-based therapies for cancer and other chronic conditions, today announced it has closed a Series C financing totalling up to $49 million. This Series C round includes an upfront investment of $32.5 million, with the right of the syndicate to invest an additional $16.5 million in the near term as programs progress. The financing was led by The Column Group and new investor Topspin Partners, and included existing investors Alta Partners, Versant Ventures, Osage Partners and ProQuest Investments. In addition, Sanofi-Genzyme BioVentures joined the syndicate as a new investor.
The funds will be used primarily to advance Immune Design’s cancer immunontherapy product pipeline, including proof-of-concept studies for its lead therapeutic candidates ID-LV305 and IDG305.
“With a focus on leveraging dendritic cell activation to fight disease, Immune Design has built on the pioneering discoveries of Dr. David Baltimore and the late Dr. Ralph Steinman to discover and develop product candidates that will hopefully result in more effective and targeted cancer treatments,” said Peter Svennilson, Founder and Managing Partner of The Column Group.
“Immune Design has made great strides in advancing its innovative immunotherapy technologies and positioning its product candidates to enter clinical evaluation,” said Bernard Davitian, Vice President and Managing Director, Sanofi-Genzyme BioVentures. “We look forward to working with the experienced team that has been assembled to help guide this company and are very pleased to support Immune Design in this financing.”
Immune Design recently expanded its management team to span all key functional areas with the addition of four veteran executives in the biotechnology and pharmaceutical fields. New team members include: Stephen Brady, Chief Business Officer; Richard T. Kenney, M.D., Chief Medical Officer; Jan Henrick ter Meulen, M.D., Chief Scientific Officer and Frank J. Hsu, M.D., Vice President, Head of Oncology. The company has also expanded operations to include a San Francisco office with plans to further expand in the Bay Area.
“With a deeply experienced leadership team and dedicated employees advancing our breakthrough molecular immunology platforms, Immune Design is committed to impacting the future of cancer treatment via our unique immunotherapy approach,” said Carlos Paya, M.D., Ph.D., President and Chief Executive Officer at Immune Design. “We are thankful for the support of our investors and hope to bring a meaningful benefit to patients in the near future.”
Immune Design’s Cancer Immunotherapy Product Candidates
• ID-LV305 is an investigational cancer therapeutic generated from the company’s DCVexTM platform, which is engineered to selectively target dermal dendritic cells in vivo to induce a powerful and durable pool of tumor- specific cytotoxic T lymphocytes (CTLs).
• ID-G305 is an investigational cancer therapeutic designed to generate tumor Ag-specific Th1 CD4 T cells, with further expansion of CTLs while catalyzing humoral and innate immune responses. ID-G305 was generated from the company’s second discovery platform leveraging TLR4 agonism, GLAASTM.
• Both ID-LV305 and ID-G305 have been engineered to target the same set of tumors expressing a specific antigen. The company plans to initiate clinical trials of these product candidates as single agents, in combination with each other as a third product candidate (ID-CMB305), and in combination with potentially complementary cancer immunotherapy approaches.
About Our New Investors
Sanofi-Genzyme BioVentures (SGBV), previously known as Genzyme Ventures, is a strategic fund that invests primarily in early-stage innovative companies in the core areas of interest of Sanofi (EURONEXT: SAN and NYSE: SNY) and its subsidiary Genzyme. SGBV’s investments focus on rare diseases, oncology, vaccines and infectious diseases, and breakthrough therapies in other core areas of interest, as well as integrated care solutions. For more information about SGBV, please visit sanofi.com or genzyme.com.
Topspin Partners is a venture capital and private equity firm that looks to generate superior returns by partnering with management to build great companies. Topspin is an affiliate of Renaissance Technologies.
About Immune Design
Immune Design is a privately-held clinical-stage biotechnology company headquartered in Seattle, WA with offices in San Francisco, CA. Immune Design brings together some of the world’s leaders in the field of molecular immunology to develop two synergistic platforms of next-generation vaccines designed to treat cancer, infectious diseases, allergy, and autoimmune disorders. The company employs leading-edge technologies that target dendritic cells for more precise activation of the immune response. These include DCVexTM, a novel lentiviral vector platform engineered to deliver antigen-encoding nucleic acids directly to dendritic cells in vivo, and GLAASTM, a TLR4 agonist platform that activates dendritic cells by up-regulating key molecules for efficient antigen presentation and produces Th1 cytokines to enhance the immune response. For more information, visit www.immunedesign.com.
# # #
Contact:
Julie Rathbun
Rathbun Communications
julie@rathbuncomm.com
206.769.9219
Cambridge, Mass., October 18, 2013 -‐-‐ Proteostasis Therapeutics, Inc., a company developing novel therapeutics that regulate protein homeostasis to improve outcomes for patients with orphan and neurodegenerative diseases, today announced the presentation of data on the Company’s lead preclinical candidates and their potential for use as combination therapies for cystic fibrosis (CF) at the 27th Annual North American Cystic Fibrosis Conference in Salt Lake City, Utah.
“These presentations demonstrate the exciting progress being made in our CF program as we work to identify a clinical candidate in 2014,” said David Weiner, M.D., Chief Medical Officer of Proteostasis Therapeutics. “Our novel Proteostasis Network (PN) platform has allowed us to identify small molecule proteostasis regulators that modulate protein homeostasis pathways to correct the folding, trafficking, and functional activity of the DF508 Cystic Fibrosis Transmembrane Regulator (CFTR). In these presentations we reported corrector activity for our compounds in human bronchial epithelial (hBE) cells, both as stand-‐ alone therapies, and in combination with existing clinical- stage corrector candidates.” The Company’s first poster and oral presentation, titled “Robust Correction of DF508 Using Small Molecule Proteostasis Regulators Alone and in Combination with Clinical-‐Stage Correctors,” were presented by Markus Haeberlein, MSc, Ph.D., Senior Vice President and Head of Research at Proteostasis Therapeutics. The Company has identified multiple novel CFTR corrector series from internal compound library screens using Multiplex Gene Expression with sentinels to pathways within the PN, and a subsequent screen for functional activity in a cystic fibrosis bronchial epithelial cell line. Identified correctors were found to have robust functional activity in DF508/DF508 hBE cells comparable to CFTR correctors currently in clinical development. Importantly, the maximal activity of those clinical-‐stage CFTR correctors was more than doubled when combined with the Company’s correctors, providing a strong foundation for the ongoing development of the program’s CFTR corrector candidates, and the potential for combination therapies.
The Company’s second poster, entitled “Correcting the DF508 Defective Folding and Trafficking Mutant of CFTR Using Small Molecule Regulators of the Proteostasis Network,” was presented by Lawrence Drew, Senior Research Associate at Proteostasis Therapeutics. The data details the Company’s broader PN-‐based approach of identifying small-‐molecule CFTR correctors, and highlights the enhanced functional activity shown by the Company’s correctors in DF508 cellular models and patient-‐derived hBE cells Compound induced modulation of PN pathways correlates with functional correction, supporting the role of PN modulation in rescuing the folding/trafficking of this disease-‐causing protein, and demonstrating proof of concept for the utilization of the PN in treating CF.
About Proteostasis Therapeutics
Proteostasis Therapeutics is developing disease-‐modifying therapeutics for orphan and neurodegenerative diseases. The Company’s lead programs in cystic fibrosis and protein aggregation diseases such as Parkinson’s disease modulate protein chaperone and proteasomal degradation pathways within the cell. These pathways are part of the cellular ‘quality control’ machinery, called the protein homeostasis network or Proteostasis Network (PN) that regulates protein folding, trafficking, and clearance. By enhancing the function and capacity of the PN, the Company’s product candidates correct for imbalances in the PN resulting from the cumulative effects of disease, genetic mutations, environmental factors, and aging. For more information, please visit www.proteostasis.com.
Investor Contact:
Jesse Baumgartner
Stern Investor Relations, Inc.
212-‐362-‐1200
SAN DIEGO, Oct. 4, 2013 (GLOBE NEWSWIRE) -- Fate Therapeutics, Inc. (Nasdaq:FATE), a biopharmaceutical company engaged in the discovery and development of adult stem cell modulators to treat orphan diseases, today announced the closing of its previously announced initial public offering of 7,666,667 shares of its common stock at a public offering price of $6.00 per share, including 1,000,000 shares of common stock issued upon the exercise in full by the underwriters of their option to purchase additional shares. Aggregate net proceeds to the Company, after deducting underwriting discounts and commissions and other estimated offering expenses, will be approximately $40.4 million.
Cowen and Company, LLC and BMO Capital Markets Corp. acted as joint book-running managers for the offering. Wedbush Securities Inc. acted as a co-manager.
A registration statement relating to these securities has been filed with and was declared effective by the Securities and Exchange Commission on September 30, 2013. The final prospectus relating to this offering has been filed with the Securities and Exchange Commission and copies may be obtained from Cowen and Company, LLC, c/o Broadridge Financial Services, Attention: Prospectus Department, 1155 Long Island Avenue, Edgewood, New York 11717, Telephone: 631-274-2806, Fax: 631-254-7140; or BMO Capital Markets Corp., Attention: Equity Syndicate Department, 3 Times Square, New York, NY 10036, Telephone: 800-414-3627, Email: bmoprospectus@bmo.com.
This press release shall not constitute an offer to sell or a solicitation of an offer to buy, nor shall there be any sale of these securities in any state or jurisdiction in which such offer, solicitation or sale would be unlawful, prior to registration or qualification under the securities laws of any such state or jurisdiction.
About Fate Therapeutics, Inc.
Fate Therapeutics is a clinical-stage biopharmaceutical company engaged in the discovery and development of pharmacologic modulators of adult stem cells to treat orphan diseases, including certain hematologic malignancies, lysosomal storage disorders and muscular dystrophies. The Company is presently advancing its lead product candidate, ProHema, a pharmacologically-modulated HSC therapeutic derived from umbilical cord blood, in Phase 2 clinical development for hematologic malignancies. Fate Therapeutics is also advancing its proprietary Wnt7a protein analogs in preclinical development for the treatment of muscular dystrophies. Fate Therapeutics is headquartered in San Diego, CA. For more information, please visit www.fatetherapeutics.com.
CONTACT: Paul Cox, Stern Investor Relations, Inc.
212.362.1200, paul@sternir.com
SOUTH SAN FRANCISCO, Calif., Sept. 26, 2013 /PRNewswire/ -- KaloBios Pharmaceuticals, Inc. (KaloBios)(NASDAQ: KBIO) today announced the pricing of an underwritten public offering of 7,500,000 shares of its common stock at a price to the public of $4.00 per share. The net offering proceeds to KaloBios are expected to be approximately $27,855,000 million, after deducting underwriting discounts and commissions and other estimated offering expenses payable by the company, but excluding any exercise of the underwriters' over-allotment option.
(Logo: http://photos.prnewswire.com/prnh/20130225/MM66380LOGO)
KaloBios anticipates using the net proceeds from this offering to develop and advance its product candidates through clinical trials, as well as for working capital and other general corporate purposes. The offering is expected to close on or about October 1, 2013, subject to customary closing conditions. In addition, KaloBios has granted the underwriters a 30-day option to purchase up to an additional 1,125,000 shares of common stock on the same terms and conditions, solely to cover overallotments, if any.
Leerink Swann LLC is acting as the sole book-running manager for the offering. William Blair & Company, L.L.C., Needham & Company LLC, and JMP Securities LLC are acting as co-managers for the offering.
The shares described above will be issued pursuant to a shelf registration statement on Form S-3 previously filed with and declared effective by the Securities and Exchange Commission (SEC) and a prospectus supplement thereto has been filed with the SEC. The offering may be made only by means of a prospectus supplement and the accompanying prospectus, copies of which may be obtained by sending a request to: Leerink Swann LLC, Attention: Syndicate Department, One Federal Street, 37th Floor, Boston, MA 02110, or via telephone at (800) 808-7525. KaloBios intends to file a final prospectus supplement relating to the offering with the SEC, which will be available along with the accompanying prospectus filed with the SEC in connection with the shelf registration, on the SEC's website at www.sec.gov.
This press release does not constitute an offer to sell or a solicitation of an offer to buy any securities of KaloBios, nor shall there be any sale of securities in any state or jurisdiction in which such an offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state or jurisdiction. This press release is being issued pursuant to and in accordance with Rule 134 under the Securities Act of 1933, as amended.
About KaloBios Pharmaceuticals, Inc.
KaloBios Pharmaceuticals, Inc. is developing a portfolio of patient-targeted, first-in-class monoclonal antibodies to treat severe medical conditions with a primary clinical focus on respiratory diseases and cancer.
Cautionary Statement Regarding Forward-Looking Statements
This communication includes forward-looking statements regarding the completion and timing of the public offering and expected use of proceeds from the public offering that involve risks and uncertainties, including, without limitation, risks related to the satisfaction of customary closing conditions. Such statements involve known and unknown risks that relate to future events or future financial performance and the actual results could differ materially from those discussed in this communication. There can be no assurance that KaloBios will be able to complete the public offering on the anticipated terms, or at all. Risks and uncertainties that may cause KaloBios's actual results to differ materially from those discussed in this communication can be found in the "Risk Factors" section of KaloBios's preliminary prospectus supplement related to the public offering filed with the SEC on September 23, 2013, and in the prospectus supplement related to the public offering that KaloBios expects to file
with the SEC on or about September 26, 2013, and in its Form 10-K, Forms 10-Q and other filings with the SEC, each available at the SEC's website at www.sec.gov. Readers are cautioned not to place undue reliance on these forward-looking statements that speak only as of the date hereof, and KaloBios assumes no responsibility to update or revise any forward-looking statements contained in this communication to reflect events, trends or circumstances after the date of this communication.
Contact:
Jeffrey H. Cooper
Chief Financial Officer
KaloBios Pharmaceuticals, Inc.
(650) 243-3146
ir@kalobios.com
Media Contact:
Joan E. Kureczka
Kureczka/Martin Associates
Tel: (415) 821-2413
Mobile: (415) 690-0210
Joan@Kureczka-Martin.com
SOURCE KaloBios Pharmaceuticals, Inc.
News Provided by Acquire Media
GAITHERSBURG, MD, September 13, 2013 – GlycoMimetics, Inc. announced today that the
the European Medicines Agency (EMA) has granted orphan drug designation for GMI-1070
(rivipansel sodium) for the treatment of vaso-occlusive crisis (VOC) in patients with sickle cell
disease. Orphan drug designation in the European Union (EU) is given to products that are
designed for the diagnosis, prevention or treatment of rare diseases that are life-threatening
or very serious. A disease is defined as rare in the EU if it affects fewer than five in 10,000
people. Granting of orphan drug designation in the EU provides companies with development
and commercial incentives, including a 10-year period of market exclusivity, access to a
centralized review process, protocol assistance and scientific advice during product
development, waiving or reduction of certain fees, and eligibility for grants and R&D support
initiatives.
“We are extremely pleased that the EMA has granted orphan drug status to GMI-1070 for
patients with sickle cell disease experiencing VOC,” said Helen Thackray, M.D., Vice President
of Clinical Development and Chief Medical Officer at GlycoMimetics. “Given that people living
with sickle cell disease currently do not have enough treatment options, we appreciate all
regulatory efforts to help bring the drug to market quickly, assuming that additional studies
will continue to demonstrate the potential for GMI-1070 to benefit those living with the
disease.”
GMI-1070, which has previously received both Orphan Drug and Fast Track status for VOC
from the U.S. Food & Drug Administration (FDA), is being developed in partnership with Pfizer
(NYSE: PFE). Pfizer is responsible for the next steps of clinical development for GMI-1070.
About GlycoMimetics, Inc.
GlycoMimetics is a clinical stage biotechnology company focused on the discovery and
development of novel glycomimetic drugs to address unmet medical needs resulting from
diseases in which carbohydrate biology plays a key role. Glycomimetics are molecules that
mimic the structure of carbohydrates involved in important biological processes. Using its
expertise in carbohydrate chemistry and knowledge of carbohydrate biology, the company is
developing a pipeline of glycomimetic drug candidates that inhibit disease-related functions of
carbohydrates, such as the roles they play in inflammation, cancer and infection. For additional
information, please visit the company’s web site at http://www.glycomimetics.com.
About Sickle Cell Disease and VOC
Sickle cell disease is a prevalent genetic disorder worldwide. Due to increasing global
migration, the number of sickle cell carriers in European countries is increasing steadily. It is a
chronic condition causing substantial illness and death. The main clinical feature of sickle cell
disease is periodic painful vaso-occlusive crisis episodes, known as VOC or pain crises, which
result in clinical complications, interruptions in patients’ lives, and cumulative irreversible
damage that impacts the morbidity and mortality of patients. Treatment for VOC today
consists primarily of supportive therapy, in the form of hydration and pain control, typically
requiring extended hospitalization.
GMI-1070 is intended to treat VOC by inhibiting the cell activation and enhanced cell adhesion
that causes the ischemia and pain. GlycoMimetics selected VOC of sickle cell disease as the
first potential indication for GMI-1070.
Contact: Brian Hahn
Email: bhahn@glycomimetics.com
Phone: 240-243-1207

CAMBRIDGE, Mass.-‐-‐(BUSINESS WIRE)-‐-‐Jun. 24, 2013-‐-‐ bluebird bio, Inc., a clinical-‐stage biotechnology company focused on transforming the lives of patients with severe genetic and orphan diseases using gene therapy, today announced the closing of its initial public offering of 6,832,352 shares of common stock at an initial public offering price of $17.00 per share, which includes the exercise in full by the underwriters of their option to purchase up to 891,176 additional shares of common stock. All of the shares in the offering were offered by bluebird bio. The company’s common stock is listed on The NASDAQ Global Select Market under the trading symbol “BLUE.”
J.P. Morgan Securities LLC and BofA Merrill Lynch are acting as joint book-‐running managers for the offering. Cowen and Company is acting as lead manager and Canaccord Genuity Inc. and Wedbush PacGrow Life Sciences are acting as co-‐managers.
A registration statement relating to these securities was declared effective by the Securities and Exchange Commission on June 18, 2013. Copies of the final prospectus relating to this offering may be obtained from J.P. Morgan Securities LLC, Attention: Broadridge Financial Solutions, 1155 Long Island Avenue, Edgewood, New York 11717, or by telephone at (866) 803-‐9204, or from BofA Merrill Lynch, 222 Broadway, New York, NY 10038, Attn: Prospectus Department, or via email, at dg.prospectus_requests@baml.com.
This press release shall not constitute an offer to sell or the solicitation of an offer to buy, nor shall there be any sale of, these securities in any state or jurisdiction in which such offer, solicitation or sale would be unlawful prior to the registration or qualification under the securities laws of such state or jurisdiction.
Source:
bluebird bio, Inc.
Investor Relations:
bluebird bio, Inc.
Richard E. T. Smith, Ph.D., 617-‐588-‐3321
rsmith@bluebirdbio.com
or
Media Contact:
Pure Communications, Inc.
Dan Budwick, 973-‐271-‐6085
dan@purecommunicationsinc.com
CHICAGO, April 23, 2013 /PRNewswire/ --
Sanofi Pasteur, the vaccines division of Sanofi (EURONEXT: SAN and NYSE: SNY), and KaloBios Pharmaceuticals (Nasdaq: KBIO) announced today that the U.S. Food and Drug Administration (FDA) has granted Fast Track designation to Sanofi Pasteur for the investigation of KB001A, an antibody fragment, intended for protection against bacterial pneumonia caused by Pseudomonas aeruginosa (Pa) in mechanically-ventilated patients. The Fast Track Drug Development Program of the FDA is designed to facilitate the clinical development and expedite the review of new drugs and vaccines that are intended to treat or prevent serious or life-threatening conditions and demonstrate the potential to address unmet medical needs. The joint announcement was made on this second day of the BIO International Convention, the 20th annual meeting of the world's largest biotechnology organization.
(Logo: http://photos.prnewswire.com/prnh/20130423/610748-a )
(Logo: http://photos.prnewswire.com/prnh/20130423/610748-b )
Most serious Pa infections occur in hospitalized and critically or chronically ill patients--primarily affecting the respiratory system in susceptible individuals--and are a serious clinical problem due to the bacteria's resistance to antibiotics. Sanofi Pasteur, which is responsible for the clinical development under the terms of the agreement with KaloBios, is currently conducting a phase I trial of the monoclonal antibody in the United States and has started the planning of a phase IIb study.
"Sanofi Pasteur is currently targeting the antibody for use in primary prevention of Pa-associated pneumonia in mechanically ventilated patients in hospitals and we are also interested in providing prevention of relapses and improvement of treatment outcomes in patients with an ongoing Pa infection," according to Michel DeWilde, Ph.D., Senior Vice President, Research and Development. "Additional indications may be considered later in the lifecycle of the product."
Under the terms of the current agreement, Sanofi Pasteur has worldwide rights to KaloBios' KB001A technology for all disease indications related to Pa infections, except cystic fibrosis and bronchiectasis, the rights in which were retained by KaloBios, and Sanofi Pasteur has the option to obtain at a later date.
"Hospital-based pneumonias, especially those associated with mechanically ventilated patients in the ICU, are a life-threatening complication that can significantly increase mortality and morbidity as well as add tens of thousands of dollars to the cost of a hospital stay," said David Pritchard, President and CEO, KaloBios. "KB001's novel mechanism of action against Pa may provide a unique means of fighting these infections, which are often resistant to antibiotic therapies. The FDA fast-track designation recognizes that this novel biologic could address an important unmet medical need."
About KB001A
KB001A, a Humaneered™ antibody fragment, is designed to fight Peudomonas aeruginosa (Pa) by blocking a virulence mechanism (the Type Three Secretion System or TTSS) on the bacterium's external surface that enables Pa to evade human immune defenses by killing white blood cells and epithelial cells, and triggering tissue- damaging inflammation. By blocking Pa's killing mechanism, KB001A is intended to reduce the damage done to the lungs by Pa and potentially enable the patient's own immune system to effectively fight and clear the bacteria from sites of infection. KB001A avoids known mechanisms of antibiotic resistance and does not contribute to broad-spectrum resistance, making it optimal for use as a single agent or in synergy with antibiotics in the preventive or therapeutic setting. KB001, a precursor, has completed two clinical studies in mechanically ventilated patients colonized with Pa and in chronically Pa infected patients with cystic fibrosis. KaloBios is currently conducting a 180-patient phase 2 study in cystic fibrosis in the United States with KB001A.
About KaloBios
KaloBios Pharmaceuticals, Inc. is developing a portfolio of patient-targeted, first-in-class monoclonal antibodies to treat serious medical conditions with a primary clinical focus on respiratory diseases and cancer. For more information on KaloBios Pharmaceuticals, please visit our web site at http://www.kalobios.com.
About Sanofi
Sanofi, a global and diversified healthcare leader, discovers, develops and distributes therapeutic solutions focused on patients' needs. Sanofi has core strengths in the field of healthcare with seven growth platforms: diabetes solutions, human vaccines, innovative drugs, consumer healthcare, emerging markets, animal health and the new Genzyme. Sanofi is listed in Paris (EURONEXT: SAN) and in New York (NYSE: SNY).
Sanofi Pasteur, the vaccines division of Sanofi, provides more than 1 billion doses of vaccine each year, making it possible to immunize more than 500 million people across the globe. A world leader in the vaccine industry, Sanofi Pasteur offers the broadest range of vaccines protecting against 20 infectious diseases. The company's heritage, to create vaccines that protect life, dates back more than a century. Sanofi Pasteur is the largest company entirely dedicated to vaccines. Every day, the company invests more than EUR 1 million in research and development. For more information, please visit: http://www.sanofipasteur.com or http://www.sanofipasteur.us
Forward Looking Statements
This press release contains forward-looking statements as defined in the Private Securities Litigation Reform Act of 1995, as amended. Forward-looking statements are statements that are not historical facts. These statements include projections and estimates and their underlying assumptions, statements regarding plans, objectives, intentions and expectations with respect to future financial results, events, operations, services, product development and potential, and statements regarding future performance. Forward-looking statements are generally identified by the words "expects", "anticipates", "believes", "intends", "estimates", "plans" and similar expressions. Although Sanofi's and KaloBios' management believes that the expectations reflected in such forward-looking statements are reasonable, investors are cautioned that forward-looking information and statements are subject to various risks and uncertainties, many of which are difficult to predict and generally beyond the control of Sanofi and KaloBios, that could cause actual results and developments to differ materially from those expressed in, or implied or projected by, the forward-looking information and statements. These risks and uncertainties include among other things, the uncertainties inherent in research and development, future clinical data and analysis, including post marketing, decisions by regulatory authorities, such as the FDA or the EMA, regarding whether and when to approve any drug, device or biological application that may be filed for any such product candidates as well as their decisions regarding labeling and other matters that could affect the availability or commercial potential of such product candidates, the absence of guarantee that the product candidates if approved will be commercially successful, the future approval and commercial success of therapeutic alternatives, the Group's or KaloBios' ability to benefit from external growth opportunities, trends in exchange rates and prevailing interest rates, the impact of cost containment policies and subsequent changes thereto, the average number of shares outstanding as well as those discussed or identified in the public filings with the SEC and the AMF made by Sanofi or the SEC made by KaloBios, including those listed under"Risk Factors" and "Cautionary Statement Regarding Forward-Looking Statements" in Sanofi's annual report on Form 20-F for the year ended December 31, 2012 and in corresponding sections of KaloBios' annual report on Form 10-K for the year ended December 31, 2012. Other than as required by applicable law, neither Sanofi nor KaloBios undertakes any obligation to update or revise any forward-looking information or statements.
KaloBios Contact: Media Contact:
Jeffrey H. Cooper Joan E. Kureczka
Chief Financial Officer Kureczka/Martin Associates
KaloBios Pharmaceuticals, Inc. Tel: +1-(415)-821-2413
+1-(650)-243-3146 Mobile: +1-(415)-690-0210
ir@kalobios.com Joan@Kureczka-Martin.com
Sanofi Pasteur Contacts:
Global Media Relations U.S. Media Relations
Pascal Barollier Susan Watkins
T. +33-4-37-37-50-38 T. +1-570-957-2563
pascal.barollier@sanofipasteur.com susan.watkins@sanofipasteur.com
http://www.sanofipasteur.com
http://www.sanofipasteur.us
SOURCE Sanofi Pasteur
News Provided by Acquire Media
San Diego, CA – Fate Therapeutics, Inc., a biopharmaceutical company engaged in the discovery and development of adult stem cell modulators, announced today the presentation of preclinical data from its WNT7a protein analog program for the treatment of muscular dystrophy at the Muscular Dystrophy Association (MDA) 2013 Scientific Conference, April 21-24, in Washington DC. The presentations describe the engineering of pharmaceutically optimized WNT7a protein analogs, as well as their mechanism of action and efficacy profile in preclinical pharmacology studies.
“The data presented today provide strong preclinical support for the therapeutic potential of WNT7a analogs in
muscular dystrophy, a complex group of disorders with a large unmet need for novel, differentiated and potentially complementary treatment approaches,” commented Christian Weyer, M.D., M.A.S., President and Chief Executive Officer of Fate Therapeutics. “We are working towards the nomination of an investigational new drug (IND) candidate later this year, and are excited about the potential to advance this new mechanism toward clinical studies.”
In the MDX mouse model of muscular dystrophy, intramuscular injection of a novel WNT7a analog resulted in
significant dose dependent muscle hypertrophy and several-fold expansion of the satellite stem cell population. Moreover, three weeks after a single intramuscular injection, functional assessment revealed a significant increase in strength of the targeted tibialis anterior muscle (+18%, p<0.001). These functional improvements were accompanied by a significant reduction in markers of fiber necrosis and inflammation typically seen in dystrophic muscle. Significant muscle hypertrophy and satellite stem cell expansion were also observed in wild-type mice. The WNT7a analogs assessed in these studies were engineered to overcome several key challenges of Wnt-family protein manufacture and formulation while retaining full biological activity and specificity. These and other data suggestthe potential of WNT7a-based protein therapeutics across a range of neuromuscular diseases and conditions.
The findings obtained with Fate’s optimized WNT7a analogs expand upon those previously reported with non-modified WNT7a protein. In November 2012, muscle biology expert and Fate Therapeutics scientific founder Dr. Michael Rudnicki published data demonstrating the potential of WNT7a to ameliorate muscle degeneration in the MDX mouse model of muscular dystrophy (Von Maltzahn et. al., PNAS 2012). In previous studies, Dr. Rudnicki elucidated theunique biology of WNT7a and its dual mechanism of action of driving the expansion of themuscle satellite stem cell population and muscle hypertrophy.
About Muscular Dystrophy
Muscular dystrophies encompass a group of disorders with diverse pathophysiological manifestations resulting from genetic aberrations which include mutations or deletions to over 30 distinct genes. The most prevalent and well characterized is Duchenne muscular dystrophy (DMD), an X-linked form of muscular dystrophy which is seen in 1/3500 live male births. DMD typically manifests in early childhood and progresses to an advanced stage of severe muscular degeneration resulting in impairment of ambulation and premature mortality. A core pathophysiologic phenomenon seen in muscular dystrophy is a cycle of muscle degeneration leading to continuous compensatory satellite cell activation and differentiation to affect a regenerative response, but resulting in the eventual exhaustion of the regenerative capacity and significant loss of muscle function. Enhancing the underlying molecular and cellular mechanisms to restore the regenerative capacity of muscle satellite stem cells thus represents a promising and unique approach for therapeutically intervention in various forms of muscular dystrophy as well as other neuromuscular diseases.
About Fate Therapeutics, Inc.
Uniquely positioned at the intersection of stem cell science and orphan disease, Fate Therapeutics is pioneering the discovery and development of innovative adult stem cell modulator therapeutics with the potential to cure or transform the lives of patients with rare life-threatening disorders. The Company’s lead program, ProHema, an innovative cord blood-derived cell therapy containing ex-vivo pharmacologically-modulated hematopoietic stem cells (HSCs), is currently in Phase 2 testing in patients with leukemia undergoing hematopoietic transplantation. The Company plans to pursue clinical evaluation of pharmacologically modulated HSCs in patients with rare genetic disorders, an area of tremendous unmet medical need in which the curativepotential of cord blood transplantation is well recognized. In addition, Fate Therapeutics is developing proprietary WNT7a-based protein therapeutics that have shown efficacy in preclinical models of muscular dystrophy. To advance its discovery efforts, the Company applies its award-winning, proprietary,induced pluripotent stem cell (iPSC) technology to generate rare cell populations and model disease. Fate Therapeutics is headquartered in San Diego, CA, with a subsidiary in Ottawa, Canada. For more information, please visit www.fatetherapeutics.com.
Contact:
Paul Cox, Stern Investor Relations, Inc.
212.362.1200, paul@sternir.com
GAITHERSBURG, MD, April 16, 2013 – GlycoMimetics, Inc. (GMI) today announced top line clinical results from a Phase 2 study of GMI-1070 in patients experiencing vasoocclusive crisis (VOC) of sickle cell disease. This randomized, double-blind, placebocontrolled trial examined the efficacy, safety and pharmacokinetics of GMI-1070, GlycoMimetics’ lead clinical-stage drug, in hospitalized patients with sickle cell disease experiencing VOC. GlycoMimetics enrolled 76 patients 12 to 60 years of age at 22 trial sites in the United States and Canada.
Patients treated with GMI-1070 experienced reduction in duration of vaso-occlusive crisis, reduction in length of hospital stay, and reduction in the use of narcotic pain relief. Both adult and pediatric patients demonstrated improvement, and adverse event rates were comparable between GMI-1070 and placebo. The company plans to submit detailed study results for presentation at a scientific meeting and publication in a peer-reviewed scientific journal.
“We are very pleased with the results of this Phase 2 study, and particularly want to acknowledge the patients and medical centers whose participation made this clinical trial possible,” said Helen Thackray, M.D., Vice President of Clinical Development and Chief Medical Officer at GlycoMimetics, Inc. “There is major unmet clinical need in sickle cell disease, and we hope that additional studies will continue to demonstrate the potential
for GMI-1070 to benefit people living with the disease.”
“This clinical data provides an important validation of GlycoMimetics’ innovative platform technology and of our approach to targeting the selectin family of adhesion molecules,”
added John Magnani, Ph.D., the company’s Vice President of Research and Chief Scientific Officer. “It is also supportive of the potential of other programs in the GlycoMimetics pipeline.”
In 2011, GlycoMimetics entered into a worldwide license agreement with Pfizer Inc. (NYSE: PFE) to develop and, if approved by applicable regulatory authorities, to commercialize GMI-1070 for all indications. Pfizer will be responsible for the next steps of clinical development for GMI-1070.
About GlycoMimetics, Inc.
GlycoMimetics is a privately held biotechnology company that capitalizes on advances in the field of glycobiology to treat inflammation, cancer, and infectious diseases. The company uses rational design of small molecule drugs that mimic the functions of bioactive carbohydrates to develop new drug candidates. For additional information, please visit the company’s web site: http://www.glycomimetics.com.
About Sickle Cell Disease and VOC
Sickle cell disease is one of the most prevalent genetic disorders in the U.S., affecting over 80,000 people. It is a chronic condition causing substantial illness and death. The main clinical feature of sickle cell disease is periodic painful vaso-occlusive crisis episodes, known as VOC or pain crises, which result in clinical complications, interruptions in patients’ lives, and cumulative irreversible damage that impacts the morbidity and mortality of patients. VOC is responsible for more than 75,000 hospitalizations per year in the U.S. with an average stay of approximately six days. Treatment for VOC today consists primarily of supportive therapy, in the form of hydration and pain control, typically requiring extended hospitalization. GMI-1070 is intended to treat VOC by inhibiting the cell activation and enhanced cell adhesion that causes the ischemia and pain. GlycoMimetics selected vaso-occlusive crisis of sickle cell disease as the first potential indication for GMI-1070.
CAMBRIDGE, MA, December 2, 2013 – bluebird bio, Inc. (Nasdaq: BLUE) a clinical-stage company committed to developing potentially transformative gene therapies for severe genetic and orphan diseases, today announced that the first subject with beta-thalassemia major has been enrolled in its phase 1/2 HGB-205 study in France and has undergone infusion with bluebird bio’s LentiGlobin drug product in an autologous hematopoietic stem cell transplantation.
“We believe gene therapy represents a potentially new and exciting treatment option for patients with severe forms of beta-thalassemia and sickle cell disease,” stated Marina Cavazzana, MD, Professor of Medicine at Paris Descartes University and Research Director at the Centre for Clinical Research in Biotherapy, Necker Hospital, Paris France. “The beta-hemoglobinopathies are the most prevalent inherited disorders worldwide, and they result in substantial morbidity and mortality. The HGB-205 study builds on early and encouraging clinical data from the LG001 study based on the pioneering work of Professor Philippe Leboulch and colleagues at the Universities of Paris and Harvard, Inserm and the French Atomic Energy and Alternative Energies Commission (CEA) research center in Paris.”
“It is very gratifying for our research, manufacturing and development teams to see their efforts to improve the potency and scalability of our product platform finally reach the clinic for patients with this life threatening disease. This milestone brings us closer towards realizing our vision of making hope a reality for patients with limited therapeutic options,” stated Dave Davidson, MD, bluebird bio’s Chief Medical Officer. “
About the HGB-205 Study
The phase 1/2 study is designed to evaluate the safety and efficacy of LentiGlobin drug product in the treatment of subjects with beta-thalassemia major and severe sickle cell disease. The study is designed to enroll up to seven subjects. Subjects will be followed to evaluate safety and transfusion requirements post-transplant. In sickle cell disease patients only, efficacy will also be measured based on the number of vaso-occlusive crises or acute chest syndrome events.
About beta-thalassemia major and sickle cell disease
Beta-thalassemia major is a rare hereditary blood disorder caused by a genetic abnormality of the beta globin gene resulting in defective red blood cells. Symptoms of beta-thalassemia include severe anemia, splenomegaly and iron overload in major organs. It is estimated that about 288,000 patients with beta-thalassemia major are alive, of which an estimated 15,000 live in the United States and Europe.
Sickle cell disease (SCD) is also a hereditary blood disorder resulting from a mutation in the beta globin gene that causes polymerization of hemoglobin proteins and abnormal red blood cell function. The symptoms of SCD include anemia, vaso-occlusive crisis and strokes. The global incidence of SCD is estimated to be 250,000 to 300,000 births annually, and the global prevalence of the disease is estimated to be about 20 to 25 million.
About bluebird bio, Inc.
bluebird bio is a clinical-stage company committed to developing potentially transformative gene therapies for severe genetic and orphan diseases. bluebird bio has two clinical-stage programs in development. The most advanced product candidate, Lenti-D, is in a recently-initiated phase 2/3 study for the treatment of childhood cerebral adrenoleukodystrophy (CCALD), a rare, hereditary neurological disorder affecting young boys. The next most advanced product candidate, LentiGlobin, is currently in a phase 1/2 study in France for the treatment of beta-thalassemia major and severe sickle cell disease. A second phase 1/2 study with LentiGlobin in the United States has been initiated for the treatment of beta-thalassemia major.
bluebird bio also has an early-stage chimeric antigen receptor-modified T cell (CAR-T) program for oncology in partnership with Celgene Corporation.
bluebird bio has operations in Cambridge, Massachusetts and Paris, France. For more information, please visit www.bluebirdbio.com
Forward-Looking Statements
This release contains “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995, including statements regarding the advancement of the Company’s clinical studies and the potential safety and clinical benefits of the Company’s product candidates. Any forward-looking statements in this press release are based on management's current expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those set forth in or implied by such forward-looking statements. These risks and uncertainties include, but are not limited to, the risk of cessation or delay of any of the ongoing or planned clinical studies and/or our development of our product candidates, the risk of a delay in the enrollment of patients in the Company’s clinical studies, the risk that the results of previously conducted studies involving similar product candidates will not be repeated or observed in ongoing or future studies involving current product candidates, the risk that our collaboration with Celgene will not continue or will not be successful, and the risk that any one or more of our product candidates will not be successfully developed and commercialized. For a discussion of other risks and uncertainties, and other important factors, any of which could cause our actual results to differ from those contained in the forward-looking statements, see the section entitled "Risk Factors" in our most recent quarterly report on Form 10-Q, as well as discussions of potential risks, uncertainties, and other important factors in our subsequent filings with the Securities and Exchange Commission. All information in this press release is as of the date of the release, and bluebird bio undertakes no duty to update this information unless required by law.
Investor Relations:
Richard E. T. Smith, Ph.D.
bluebird bio, Inc
(617) 588-3321
Media Contact:
Dan Budwick
Pure Communications, Inc.
(973) 271-6085
NOVATO, CA – December 19, 2012 -‐ Ultragenyx Pharmaceutical Inc., a biotechnology company focused on the development of treatments for rare and ultra-‐rare genetic disorders, today announced the successful completion of a $75 million Series B round financing led by Adage Capital Partners, LP. Joining Adage as new investors in this financing are mutual funds and separate account clients advised by T. Rowe Price Associates, Inc., Jennison Associates LLC (on behalf of clients), funds and accounts under management by subsidiaries of BlackRock, Inc., Sanofi-‐Genzyme BioVentures¹, Shire plc and additional blue chip public market funds. Existing investors, TPG Biotech, Fidelity Biosciences, HealthCap and Pappas Ventures also participated in the transaction.
Ultragenyx plans to use the proceeds from the financing primarily to advance development of the company’s lead clinical-‐stage programs, UX001 and UX003, and other undisclosed programs. UX001 is a potential substrate replacement therapy for hereditary inclusion body myopathy currently being investigated in a fully enrolled, randomized placebo-‐controlled Phase 2 clinical study with results anticipated in 2013. UX003 is a recombinant enzyme replacement therapy intended for the treatment of mucopolysaccharidosis type 7 (MPS 7), which will enter a Phase 1/2 clinical study in MPS 7 patients in 2013. The company intends to continue to expand its pipeline through the potential in-‐licensing of additional products.
“We welcome the participation of such highly regarded public market and strategic investors in this pivotal financing,” said Eran Nadav, PhD, Managing Director at TPG Biotech and Chairman of the Board of Ultragenyx. “Ultragenyx, led by a distinguished team experienced in rare disease drug development, is building a world-‐class orphan drug company with a rich pipeline of transformative therapeutics.”
1h has be med to reflect an expanded strategic investment focus on areas of interest to both Sanofi and Genzyme.
Transforming good science into great medicine for rare genetic diseases
www.ultragenyx.com
“We deeply appreciate the support of all our investors, new and existing, and their confidence in our ability to find and efficiently develop compelling new treatments for devastating rare genetic
disorders,” said Emil D. Kakkis, MD, PhD, Chief Executive Officer and Founder of Ultragenyx. “This financing transaction is critical to expanding our efforts to deliver profound novel therapies that
benefit even more rare disease patients.”
Cowen and Company served as financial advisor on the financing. Ropes & Gray LLP served as legal advisor.
About Ultragenyx
Ultragenyx is a privately held, development-‐stage biotechnology company committed to bringing to market life-‐transforming therapeutics for patients with rare and ultra-‐rare metabolic genetic diseases. The company focuses on diseases for which the unmet medical need is high, the biology for treatment is clear, and for which there are no effective treatments.
The company is led by a management team experienced in the development and commercialization of rare disease therapeutics. Ultragenyx’ strategy is predicated upon time and cost-‐efficient drug development, with the goal of delivering safe and effective therapies to patients with the utmost urgency.
For more information on Ultragenyx, please visit the company’s website at www.ultragenyx.com.